

购物车
 您的购物车当前为空
您的购物车当前为空

 很棒
很棒GDC-2992是一种选择性和口服的AR (androgen receptor) 降解剂,在VCaP细胞中降解AR(DC50=2.7 nM)和抑制增殖(IC50=9.7 nM),可用于研究去势抵抗性前列腺癌(CRPC)。
很棒纯度: 99.82%
别名 RO7656594, RO 7656594, GDC2992
GDC-2992是一种选择性和口服的AR (androgen receptor) 降解剂,在VCaP细胞中降解AR(DC50=2.7 nM)和抑制增殖(IC50=9.7 nM),可用于研究去势抵抗性前列腺癌(CRPC)。
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1 mg | ¥ 2,880 | 现货 | |
| 5 mg | ¥ 6,570 | 现货 | |
| 10 mg | 待询 | 现货 | |
| 25 mg | 待询 | 现货 | |
| 50 mg | 待询 | 待询 |
TargetMol的所有产品仅用作科学研究或药证申报,不能被用于人体,我们不向个人提供产品和服务。请您遵守承诺用途,不得违反法律法规规定用于任何其他用途。
凭借在化合物合成方面的丰富经验,我们可以根据您的研究需求为该产品提供快速定制合成服务。
| 产品描述 | GDC-2992 is a selective and orally active androgen receptor (AR) degradator that degrades AR (DC50 = 2.7 nM) and inhibits proliferation (IC50 = 9.7 nM) in VCaP cells, making it suitable for studying castration-resistant prostate cancer (CRPC). |
| 靶点活性 | EGFR (L858R):1.1 nM, EGFR (WT): 5.6 nM |
| 体外活性 | GDC-2992 在多种 EGFR 突变细胞模型中表现出强效抑制活性,特别是对 EGFR L858R 和 T790M/L858R 突变体,IC₅₀ 分别为 1.1 nM 和 1.0 nM。在肺癌细胞系 H1975(携带 T790M/L858R 突变)中,其增殖抑制 IC₅₀ 为 5.6 nM,同时可显著抑制 p-EGFR、p-AKT 和 p-ERK 的表达,说明其对 EGFR 信号通路具有有效阻断作用。对野生型 EGFR 的抑制力较弱(IC₅₀ > 100 nM),显示出良好的突变选择性[1]。 |
| 体内活性 | 在携带 H1975 肿瘤的 NSG 小鼠模型中,GDC-2992 经口服每日给药(QD)显示出剂量依赖性的抗肿瘤作用,在 100 mg/kg 剂量下可实现肿瘤几乎完全消退。其在体内有效抑制肿瘤组织中 EGFR 磷酸化水平(p-EGFR),验证其作用机制[1]。 |
| 别名 | RO7656594, RO 7656594, GDC2992 |
| 分子量 | 819.39 |
| 分子式 | C45H51ClN8O5 |
| CAS No. | 2753651-10-2 |
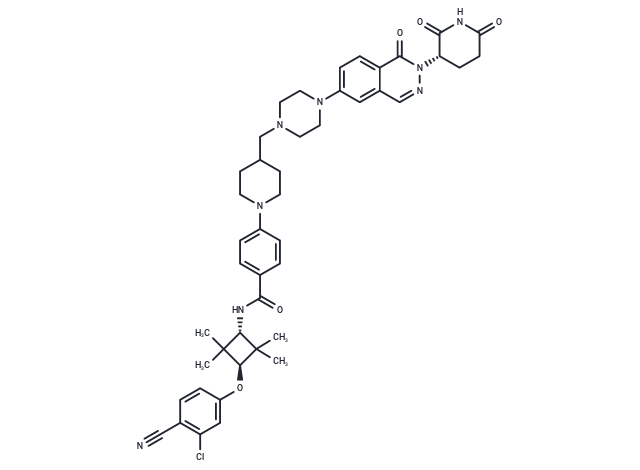
| Smiles | O([C@H]1C(C)(C)[C@H](NC(=O)C2=CC=C(C=C2)N3CCC(CN4CCN(CC4)C=5C=C6C(=CC5)C(=O)N(N=C6)[C@@H]7C(=O)NC(=O)CC7)CC3)C1(C)C)C8=CC(Cl)=C(C#N)C=C8 |
| 密度 | no data available |
| 存储 | Keep away from moisture Powder: -20°C for 3 years | In solvent: -80°C for 1 year Shipping with blue ice/Shipping at ambient temperature. 实际储存温度请以COA为准 | |||||||||||||||||||||||||
| 溶解度信息 | DMSO: 30 mg/mL (36.61 mM), Sonication is recommended. | |||||||||||||||||||||||||
溶液配制表 | ||||||||||||||||||||||||||
DMSO
该溶液配制表仅适用于固体产品。对于液体产品,请根据标明的浓度或密度计算稀释方案。 | ||||||||||||||||||||||||||
对于不同动物的给药剂量换算,您也可以参考 更多
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询
版权所有©2015-2026 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4 | 沪公网安备 31010602006700号 | 沪(静)应急管危经许[2024]203441
| 沪(静)应急管危经许[2024]203441