购物车
 您的购物车当前为空
您的购物车当前为空

动态分析,描绘分子真实状态




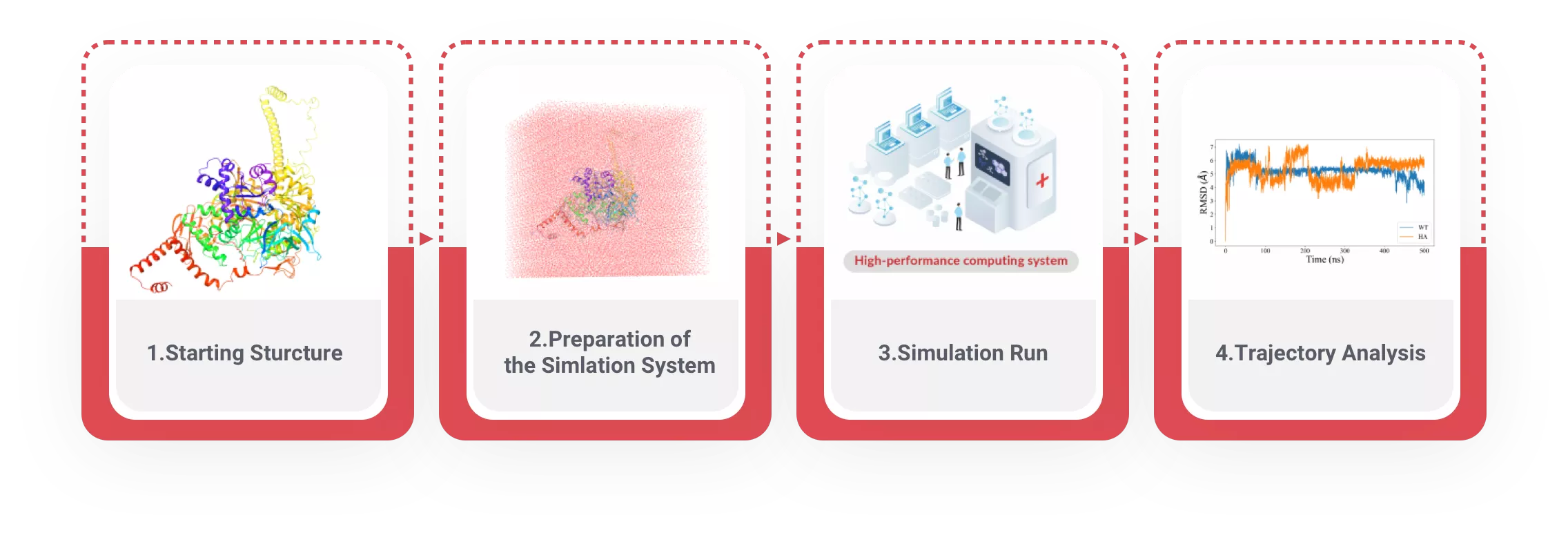
分子动力学是基于经典力学的一种分子模拟方法。与分子力学不同,分子动力学求解的是随时间变化的分子的状态、行为和过程。该方法模拟分子运动的过程,它按照分子瞬时的运动状态,求解每一个原子的牛顿运动方程和每一个原子的位置和速度,并从这一运动轨迹中计算得到各种性质。如果在所研究的模拟时间内能正确地选取分子体系的力场函数形式及其参数,分子动力学就可成为解析分子运动性质的一种强大工具。
蛋白质的结构和功能密切相关,蛋白质不同的构象对应不同的生物功能、所有的生物大分子在生理状态下都是动态的,蛋白质是动态的存在和发挥功能的,生物体内的一系列生物事件的发生是依靠蛋白质的动态构象变化来调节的,药物与蛋白结合或离解的过程也是动态变化的。蛋白质的构象变化,小到局部的残基侧链的位置变化,大到整个结构域的运动,时间尺度从飞秒(fs)到毫秒(ms)甚至秒(s)有很大的波动范围。研究蛋白质的动态性质,对进一步认识蛋白质的结构功能关系和通过结构功能关系知识进一步了解和控制生命过程具有重大意义。
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询
版权所有©2015-2026 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4 | 沪公网安备 31010602006700号 | 沪(静)应急管危经许[2024]203441
| 沪(静)应急管危经许[2024]203441