

购物车
 您的购物车当前为空
您的购物车当前为空


 还可以
还可以PLK1-IN-13 是一种具选择性且可口服的 PLK1 抑制剂 (IC50: 0.27 nM),同时抑制 PLK2 (IC50: 12.72 nM) 和 PLK3 (IC50: 4.12 nM)。它使细胞停滞于 G2 期,引发细胞凋亡 (apoptosis),并抑制与增殖相关的致癌基因 c-MYC 的转录。PLK1-IN-13 在抑制肿瘤生长方面表现出色,适用于急性髓系白血病 (AML) 研究。
还可以PLK1-IN-13 是一种具选择性且可口服的 PLK1 抑制剂 (IC50: 0.27 nM),同时抑制 PLK2 (IC50: 12.72 nM) 和 PLK3 (IC50: 4.12 nM)。它使细胞停滞于 G2 期,引发细胞凋亡 (apoptosis),并抑制与增殖相关的致癌基因 c-MYC 的转录。PLK1-IN-13 在抑制肿瘤生长方面表现出色,适用于急性髓系白血病 (AML) 研究。
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mg | 待询 | 10-14周 | |
| 50 mg | 待询 | 10-14周 |
TargetMol的所有产品仅用作科学研究或药证申报,不能被用于人体,我们不向个人提供产品和服务。请您遵守承诺用途,不得违反法律法规规定用于任何其他用途。
凭借在化合物合成方面的丰富经验,我们可以根据您的研究需求为该产品提供快速定制合成服务。
| 产品描述 | PLK1-IN-13 is a selective, orally active PLK1 inhibitor with an IC50 of 0.27 nM. It also inhibits PLK2 (IC50: 12.72 nM) and PLK3 (IC50: 4.12 nM). PLK1-IN-13 induces cell cycle arrest at the G2 phase, promotes apoptosis, and downregulates the transcription of the cancer-associated oncogene c-MYC. This compound inhibits tumor growth and is applicable for research in acute myeloid leukemia (AML). |
| 靶点活性 | PLK1:0.27 nM |
| 体外活性 | PLK1-IN-13(72 h)(Compound WD6)对 MDA-MB-231 细胞(IC 50:9.5 nM)和 MV4-11 细胞(IC 50:23.3 nM)具有抗增殖活性,而对 MCF-7 和 HCT-116 的 IC 50 分别为 47.1 nM 和 59.9 nM。该化合物对 CYP2C9(IC 50:7.39 μM)、CYP2C19(IC 50:14 μM)、CYP2D6(IC 50:13.8 μM)和 CYP3A4(IC 50:17.3 μM)的抑制作用较弱,暗示其可能具有较低的药物相互作用风险。PLK1-IN-13 在浓度 2.92 nM-46.6 nM(72 小时)下诱导 MV4-11 细胞发生 G2/M 细胞周期停滞及诱导凋亡。此外,在 0-1 μM(24 小时)条件下,该化合物下调 MV4-11 细胞中增殖相关致癌基因 c-MYC 的转录。 |
| 体内活性 | PLK1-IN-13 (Compound WD6) (20 mg/kg,口服,每天一次,连续 19 天) 在 MV4-11 异种移植小鼠模型中表现出显著的肿瘤生长抑制效果 (TGI = 91.2 %)。在 SD 大鼠中,PLK1-IN-13 (10、20 mg/kg,口服) 呈现出良好的半衰期、高血浆暴露量和中等生物利用度。 |
| 分子量 | 577.744 |
| 分子式 | C29H39N9O2S |
| CAS No. | 3038489-48-1 |
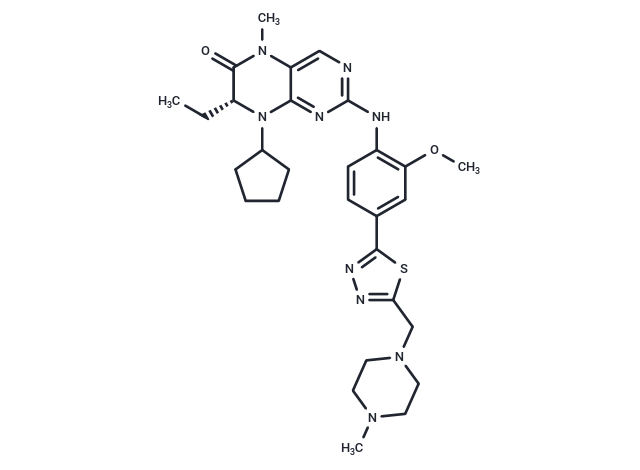
| Smiles | C(C)[C@H]1N(C=2C(N(C)C1=O)=CN=C(NC3=C(OC)C=C(C=C3)C=4SC(CN5CCN(C)CC5)=NN4)N2)C6CCCC6 |
| 存储 | Powder: -20°C for 3 years | In solvent: -80°C for 1 year Shipping with blue ice/Shipping at ambient temperature. 实际储存温度请以COA为准 |
对于不同动物的给药剂量换算,您也可以参考 更多
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询
版权所有©2015-2026 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4 | 沪公网安备 31010602006700号 | 沪(静)应急管危经许[2024]203441
| 沪(静)应急管危经许[2024]203441