
购物车
 您的购物车当前为空
您的购物车当前为空
铁死亡综述!解码铁死亡与癌症治疗

自铁死亡发现以来,越来越多的研究表明,其失调与癌症的其它标准特征相交叉,从而将其定为为一个多效靶点。然而,目前尚无基于铁死亡的获批药物,其被推崇的转化价值仍处于一种相对推测的阶段。
2025年10月10日,一篇发表于 Nature reviews cancer 期刊的论文探讨了铁死亡的生化基础、支持其在癌症生物学中作用的证据,以及合理化靶向疗法以诱导恶性肿瘤中铁死亡倾向状态的潜在策略,强化这些交叉领域的研究可能会带来基于铁死亡的创新治疗范式。跟T仔一起来快速浏览一下吧~ (私信T仔可获取原文噢)

一、铁死亡的生化基础
铁死亡首次于2012年提出,这种细胞死亡形式的特点包括铁依赖性的脂质过氧化和随后的质膜破裂。当铁水平和脂质氧化的调节出现问题时,铁死亡就会发生。
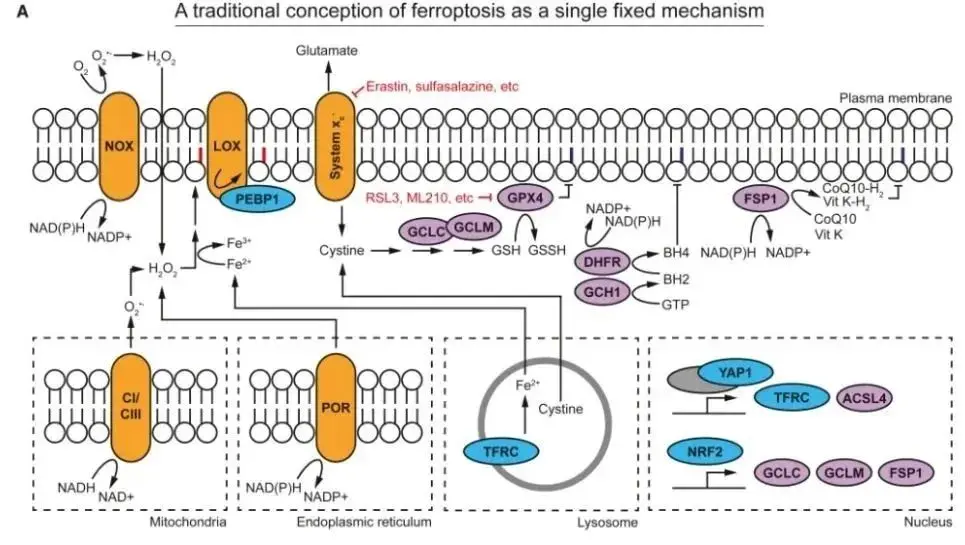
▲铁死亡传统通路图
脂质过氧化作为铁死亡的核心,涉及多不饱和脂肪酸(PUFAs)在膜磷脂中的脂质和脂质过氧自由基(·OOH)的形成,进而启动并传播自氧化反应,导致磷脂过氧化物(PL-OOHs)和脂质过氧化分解产物,如反应性醛类的形成,最终导致膜结构破裂和细胞死亡。铁在这一过程中发挥着关键作用——通过Fenton反应催化ROS生成,推动脂质氧化反应。
铁依赖性脂氧合酶(LOXs)最初被认为主导此过程,但最近的研究表明,其它氧化还原酶(如NADPH-细胞色素P450还原酶)也可通过生成ROS,进而通过芬顿反应产生·OH自由基发挥作用。
此外,铁的摄取、储存和调控路径可能影响癌细胞的铁死亡敏感性。肿瘤细胞常通过转铁蛋白受体1(TFRC)或CD44-透明质酸介导的机制摄取铁,导致铁死亡敏感性增加。KRAS突变、MYCN扩增或NRF2通路异常也会改变细胞的氧化平衡,使其更易受铁死亡影响。
该综述还提到 细胞主要依赖三大防御系统来防止铁死亡:
System xc- -GSH-GPX4 轴:通过GPX4减少磷脂过氧化物,从而避免铁死亡。药理抑制System xc–或GSH耗竭可诱导铁死亡。
FSP1–NAD(P)H–辅酶Q₁₀途径:FSP1将氧化型辅酶Q₁₀还原为还原型辅酶Q₁₀(CoQ₁₀-H₂),起到防止脂质过氧化的作用。
GCH1-BH4途径:作为备用系统,通过生成四氢生物蝶呤(BH₄)抑制脂质过氧化,但在某些血液肿瘤中,这一系统的活性升高可能限制铁死亡治疗的效果。
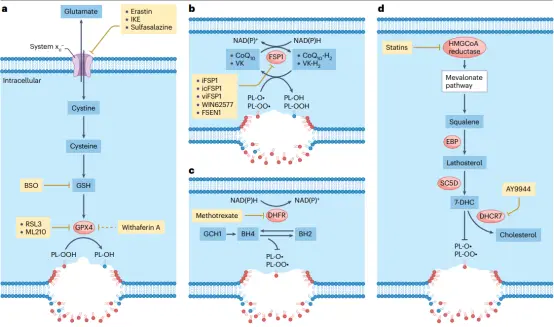
▲铁死亡监控系统及其可操作性
二、从氧化还原生物学视角看铁死亡
该综述提到,早期研究认为是 ROS(活性氧)促进基因突变、驱动癌变;但后续发现抗氧化剂可能反而加重癌症,因为它们削弱了癌细胞自我调控 ROS 的能力。癌细胞典型的代谢异常(如Warburg效应、线粒体功能障碍、氨基酸代谢重编程)显著影响其氧化还原平衡与铁死亡敏感性。
肿瘤细胞 ROS 主要来源于线粒体电子传递链泄漏及 NADPH 氧化酶(NOX)活性,这些复杂交互使得难以明确 ROS 驱动的下游靶点,因此理解铁死亡的遗传基础对于阐明其与ROS的关系至关重要。
1、NRF2通路在癌症与铁死亡中的关键作用
KEAP1–NRF2轴是癌细胞抵御ROS损伤的核心调控机制。正常情况下,KEAP1与Cullin3介导NRF2降解;当ROS累积时,KEAP1的半胱氨酸残基被修饰,NRF2被激活,引发抗氧化反应。
NRF2具有双重作用:在早期癌变中抑制肿瘤形成;在晚期癌症中,KEAP1、CUL3或NRF2突变导致NRF2持续激活,促进肿瘤存活、耐药和代谢重编程。
高NRF2活性增强抗氧化防御、抑制细胞凋亡并抑制铁死亡。例如在非小细胞肺癌(NSCLC)中,KEAP1/NRF2/STK11突变使肿瘤对铁死亡具有高度耐受性,脂质代谢关键酶SCD1和抗氧化还原酶AKR1C家族被上调。
实验表明:
抑制SCD1可增强铁死亡诱导剂效果;
• 下游分子FSP1参与NRF2介导的放射抗性和铁死亡抗性;
• 联合抑制NRF2与FSP1可协同诱导铁死亡,为克服癌症抗药性提供新策略。
2、氧化还原网络与抗氧化防御
肿瘤细胞中ROS清除依赖多条抗氧化途径,如铁代谢、TXN(硫氧还蛋白)、NADPH及谷胱甘肽(GSH)系统。其中,GSH由γ-谷氨酰半胱氨酸合成酶(γ-GCS)催化合成,是细胞内主要电子供体。
• GSH合成对肿瘤起始阶段至关重要,但在肿瘤建立后,细胞可通过TXN系统补偿;
• 早期抑制GSH合成可防止肿瘤发生,但对已形成的肿瘤作用有限;
• 同时抑制GSH与TXN通路可显著诱导癌细胞死亡,提示联合靶向的治疗潜力。
3、MYCN扩增与铁死亡易感性
在神经母细胞瘤(neuroblastoma)中,约20–25%的病例存在MYCN扩增,显著改变细胞铁与ROS代谢:
• MYCN增强铁摄取(通过TFRC)与ROS生成,增加铁死亡敏感性;
• MYCN扩增细胞依赖系统x_c⁻(SLC7A11)来解毒ROS,因此对铁死亡诱导剂极度敏感;
• 抑制半胱氨酸摄取、转硫途径或GPX4可诱导肿瘤缓解。
此外,研究发现LRP8介导硒(用于合成GPX4)的摄入,对防止铁死亡至关重要;缺失LRP8可诱导铁死亡,是潜在治疗靶点。
4、铁死亡在铁代谢和癌症中的角色
铁是多种关键酶(如LOX、COX、HIF羟化酶)的辅因子,但其自由形态可通过Fenton反应产生ROS。癌细胞表现出“铁依赖性(iron addiction)”以维持高代谢状态。
相关机制包括:
• CD44介导的铁结合透明质酸摄取(独立于转铁蛋白受体);
• NCOA4介导的铁蛋白自噬(ferritinophagy)释放游离铁,从而推动铁死亡;
• 老化相关研究表明,NUPR1–Lipocalin 2通路调控铁代谢和肺癌干性,铁补充可恢复老化细胞的致瘤潜能。
•
三、癌症代谢与铁死亡
癌症代谢研究的起点来自 Otto Warburg 在1924年提出的“癌细胞代谢改变”理论(Warburg 效应),最初认为癌变源于异常代谢,后续由于 SRC、EGFR 等癌基因的发现,研究重心转向基因突变。到如今,研究者认为 代谢重塑与致癌信号共同驱动癌症发生与发展;
1. 半胱氨酸代谢与铁死亡
• 癌细胞对半胱氨酸依赖性(cysteineaddiction)是铁死亡敏感性的核心决定因素。
• 通过降解半胱氨酸(如使用工程化cyst(e)inase)可阻断谷胱甘肽(GSH)合成与GPX4活性,诱导铁死亡。
• BAP1基因突变会失去对SLC7A11的表观遗传抑制作用,从而抑制铁死亡。
• p53可通过抑制SLC7A11增强铁死亡敏感性,但部分TP53突变型(如非洲裔人群特有突变)反而导致抗铁死亡性。
2.维生素与微生物代谢物的作用
多种维生素具备天然抗铁死亡作用:
• 维生素A、E、K为天然自由基捕获剂(RTAs);
• 维生素K循环酶VKORC1L1能清除磷脂过氧化物,被p53下调后可增强铁死亡;
• warfarin抑制VKORC1L1,从而促进铁死亡、抑制肿瘤生长;
• 氧化型维生素C(DHAA)与IKE联合能有效诱导对IKE耐药的胶质瘤细胞铁死亡。
• 肠道微生物代谢物也影响肿瘤铁死亡:
• 由厌氧消化链球菌(Peptostreptococcusanaerobius)产生的吲哚丙烯酸(IDA)抑制铁死亡、促进结直肠癌生长;其机制涉及AHR–ALDH1A3轴,产生NADH以还原CoQ₁₀并抑制脂质过氧化。
此外,铁死亡不仅导致细胞死亡,还会释放氧化脂质介质,这些分子可作为免疫信号招募中性粒细胞等免疫细胞。然而,这些信号可能既促进免疫激活,也可能诱导免疫抑制微环境,从而帮助肿瘤免疫逃逸。
四、铁死亡与癌症免疫
不同于免疫耐受性细胞死亡(如凋亡)或免疫原性死亡(如焦亡、坏死性凋亡),铁死亡可同时激活或抑制免疫反应,其免疫效应依赖死亡阶段与微环境背景。
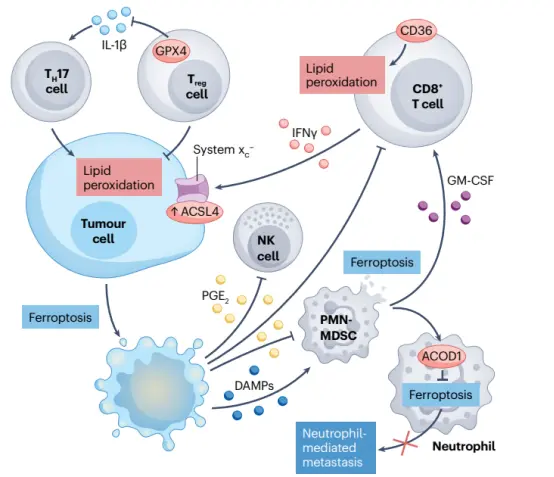
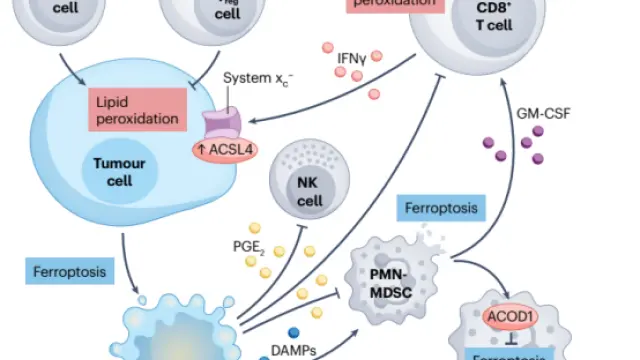
▲铁死亡视角下的肿瘤–免疫互作
1. 铁死亡与适应性免疫
铁死亡在适应性免疫中扮演着复杂的角色,尤其与 CD8⁺ T 细胞、Treg细胞、肿瘤微环境中的脂质过氧化对T细胞功能的影响密切相关:
• 免疫检查点抑制激活的CD8⁺ T细胞通过分泌IFNγ,抑制肿瘤细胞的SLC3A2/SLC7A11,减少半胱氨酸摄取,引发脂质过氧化和铁死亡,增强抗肿瘤作用。
• 花生四烯酸(AA)和ACSL4也参与铁死亡介导的T细胞抗肿瘤效应;缺乏ACSL4可加速肿瘤生长。
• GPX4在Treg细胞中维持氧化还原平衡,防止脂质过氧化和铁死亡。
• 特异性敲除Gpx4的Treg细胞产生线粒体ROS和IL-1β,激活TH17细胞,抑制肿瘤生长,提示选择性靶向Treg的GPX4可增强抗肿瘤免疫。
• 全身性抑制GPX4可能损害效应T细胞功能,需要谨慎设计治疗策略。
• 肿瘤微环境中脂质通过CD36受体进入CD8⁺ T细胞,导致脂质过氧化和p38激酶活化,引起T细胞功能障碍。
• 阻断氧化脂质–CD36轴可恢复T细胞功能、改善免疫治疗效果。
2. 铁死亡与先天免疫
铁死亡在先天免疫中同样重要。
• 铁死亡癌细胞释放前列腺素E₂(PGE₂),抑制cDC1和NK细胞活化,促进免疫逃逸,尤其在高PGE₂肿瘤中。
• 肿瘤微环境中PMN-MDSCs自发铁死亡,减少其数量,但释放的氧化脂质抑制T细胞功能。
• 抑制PMN-MDSCs铁死亡(如Liproxstatin-1)可增强免疫检查点疗效、减缓肿瘤进展;诱导铁死亡(如IKE)则促进肿瘤生长。提示:PMN-MDSC铁死亡是一种免疫抑制机制,可作为治疗靶点。
• ACOD1在肿瘤浸润中性粒细胞中受GM-CSF–JAK–STAT5–C/EBPβ通路激活,保护其免于铁死亡,支持转移。
• 靶向ACOD1可减少中性粒细胞浸润、增强抗肿瘤免疫,改善转移性乳腺癌免疫治疗效果。
五、铁死亡易感的癌细胞状态
铁死亡易感性受肿瘤微环境、细胞密度、代谢重编程及脂质组成等因素影响。其不仅受细胞自身机制控制,还受邻近细胞的直接相互作用影响。
具体体现为:
• 高细胞密度可抑制铁死亡,即使GPX4缺失。这说明实体瘤固有的高密度特性本身会产生抗铁死亡效应,并影响氧气浓度、半胱氨酸供应及细胞应激适应能力。
• 铁死亡可在细胞群体中以波浪式扩散,类似焦亡和坏死性凋亡,依赖脂质过氧化物和铁,但不完全依赖GPX4抑制。
• 上皮-间充质转化(EMT)后的肿瘤细胞因脂质代谢改变,对铁死亡更易感,尤其是膜磷脂多不饱和脂肪酸(PUFA)增加。
• 耐药持久性细胞中,脂质过氧化与铁死亡密切相关,铁死亡可成为消除这些难治细胞的潜在策略。
• 黑色素瘤通过淋巴系统转移时,由于氧化应激水平较低,更容易形成转移灶;而经血液转移则依赖于 system xc⁻–GSH–GPX4 轴维持抗氧化平衡。淋巴环境中富含谷胱甘肽(GSH)和单不饱和脂肪酸(MUFA,如油酸),可诱导细胞进入抗铁死亡状态。
• 体外或尾静脉注射实验显示,删除SLC7A11可降低黑色素瘤的转移能力,提示淋巴环境通过降低铁死亡促进转移。
激活铁死亡的挑战与策略
铁死亡在癌症治疗中的应用被视为一把“双刃剑”,一方面需要防止其在正常组织中引起的过度组织损伤,另一方面又希望在恶性肿瘤中通过诱导铁死亡实现细胞清除。因此,如何精准调控铁死亡,既维持组织稳态又促进肿瘤细胞死亡,是当前临床转化中的关键难题。
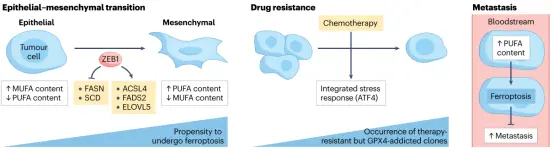
▲铁死亡对难治性癌症状态的影响
铁死亡独特的机制提供了几种治疗途径:
1. 诱导癌细胞发生铁死亡,这可以通过抑制抗氧化途径来实现(如直接抑制GPX4)。
2. 增强铁依赖性脂质过氧化。能增加细胞内铁水平的化合物,如在促铁死亡环境中的铁螯合剂或能从储存蛋白中释放铁的化合物,都能促进铁死亡(例如,系统xc⁻的抑制剂erastin)。
3. 将铁死亡诱导剂与其他抗癌方式结合。
由此也诞生了以下疗法:
靶向关键分子:开发GPX4的PROTAC降解剂(如dGPX4),通过脂质纳米颗粒(dGPX4@401-TK-12)靶向肿瘤,小鼠模型中抑制肿瘤生长且副作用小;icFSP1诱导FSP1相分离,与GPX4抑制剂联用抑制KEAP1突变肺癌生长;brequinar(DHODH抑制剂)靶向GPX4低表达肿瘤,与sulfasalazine 联用靶向高表达肿瘤。
联合治疗:抗PD-1抗体与IKE联用,增强黑色素瘤模型抗肿瘤效应;放疗激活ATM下调SLC7A11,结合CD8⁺T细胞分泌的IFNγ,协同诱导肺癌铁死亡。
优化递送:利用ultrasmall(<10 nm)PEG修饰silica纳米颗粒,搭载黑色素瘤靶向肽,在营养缺乏癌细胞中诱导铁死亡,抑制肿瘤复发。
总体而言,铁死亡在癌症中的作用和其调控机制复杂,针对铁死亡的治疗策略需要考虑铁代谢、脂质过氧化及抗氧化机制的相互作用,以期在癌症治疗中取得更好的效果。
科研助力
TargetMol可为您提供多种铁死亡抑制剂、诱导剂,以及化合物库。
TargetMol 铁死亡化合物库 ,收集了 779 种与铁死亡通路相关的化合物,靶点包括 GPX4、ROS、p53、Nrf2等,另外还包含了铁螯合剂、抗氧化剂等多种类型小分子,助力您铁死亡通路相关的靶点鉴定和药物研发。
779种与铁死亡通路相关的化合物,可用于高通量和高内涵筛选;
靶点含GPX4,ROS,p53,Nrf2等;
部分化合物已被FDA批准上市;
详细的说明书,化合物结构、靶点信息、活性描述等;
NMR、HPLC/LCMS等多种检测技术保证产品结构正确,纯度高,减少假阳性。
如需了解更多详细信息,欢迎随时私信我们~
原文链接:
https://www.nature.com/articles/s41568-025-00864-1


科学新闻、观点和分析的重要汇总,每个工作日都会发送到您的收件箱.
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询
版权所有©2015-2026 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4 | 沪公网安备 31010602006700号 | 沪(静)应急管危经许[2024]203441
| 沪(静)应急管危经许[2024]203441