购物车
 您的购物车当前为空
您的购物车当前为空
PNAS | 最新研究揭示调控线粒体自噬的新机制!

线粒体自噬(Mitophagy)是细胞选择性清除受损线粒体的过程,也是维持线粒体网络与氧化还原稳态的重要途径。其启动依赖于PINK1 (PTEN 诱导激酶 1)在线粒体外膜上的稳定积累,从而引发泛素依赖性的线粒体自噬。然而,触发线粒体自噬的起始信号一直不清楚。
一些研究关注了 mtROS 在线粒体自噬启动中的作用,但 mtROS 是否本身作为信号分子直接启动线粒体自噬,以及其下游效应因子仍不明确。
2025年9月30日,由中国医科大学曹流教授、郭启强教授、宋晓宇教授、匹兹堡大学医学院Toren Finkel教授通讯发表于 PNAS 期刊的论文揭示了由 mtROS 通过 ATM-CHK2 通路调控线粒体自噬的新机制。跟 T 仔一起来看看吧~

mtROS信号启动线粒体自噬
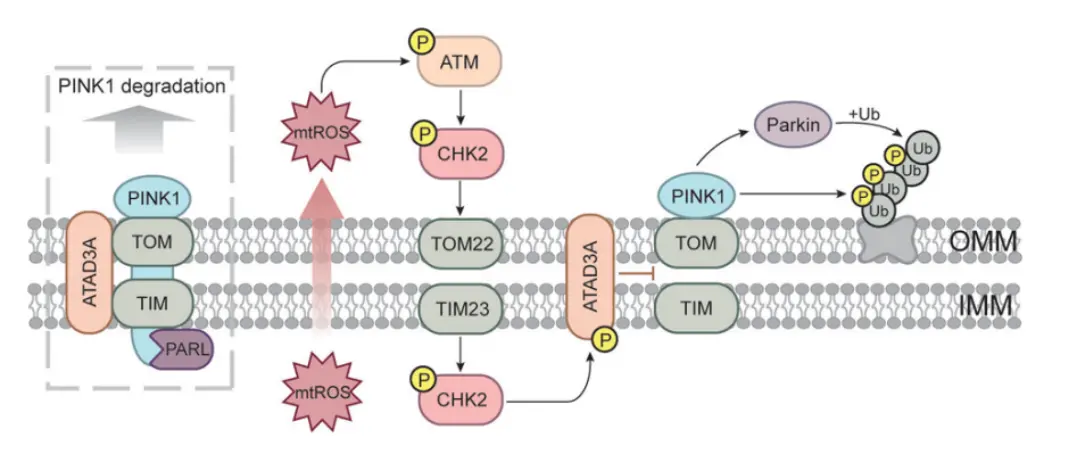
线粒体自噬的启动依赖于 PINK1 在线粒体外膜(OMM)上的稳定积累,从而引发泛素依赖性的线粒体自噬。在健康线粒体中,PINK1 前体蛋白通过外膜转运复合体(TOM)和内膜转运复合体(TIM23)部分导入线粒体,其过程由 N 端的线粒体靶向序列(MTS)引导。通过 TIM23 复合体后,PINK1 先后被线粒体加工肽酶(MPP)及 PINK1/PGAM5 相关的类菱形蛋白酶(PARL)切割。随后,PINK1 被逆向转运至胞质中,并通过 N 端规则途径经蛋白酶体持续降解。然而,当线粒体去极化发生时,PINK1 的切割被阻断,从而在受损线粒体外膜上稳定积累。此时,PINK1 招募并磷酸化 E3 泛素连接酶 Parkin 的 Ser65 位点,以及泛素(UB)分子中保守的 Ser65 位点,激活的 Parkin 随后促使受损线粒体外膜蛋白发生泛素化修饰。除去极化外,线粒体蛋白质错误折叠同样可触发 PINK1/Parkin 依赖的线粒体自噬,这一过程由前序易位酶相关运动 (PAM) 复合体介导。
在最新研究中,研究者揭示了 mtROS 可作为信号分子直接激活 ATM-CHK2 通路。被激活的 CHK2(checkpoint kinase 2)与线粒体膜蛋白 ATAD3A 相互作用,并在 Ser371 位点磷酸化该蛋白。此磷酸化事件破坏了 PINK1 与线粒体转运复合体 TOM22/TIM23 的结合,从而阻断 PINK1 向内膜的转运,使 PINK1 在外膜积累并触发 PINK1/Parkin 依赖的线粒体自噬。
高通量筛选示意图
ATAD3A 是 PINK1 运输与加工的桥接因子。当细胞受到抗氧剂处理时,ATAD3A 与 TIM23 以及 TIM23 与 PINK1 的相互作用均被破坏。然而,究竟是哪种信号破坏了 ATAD3A 与 TIM23 的结合机制此前并不清楚。
这项研究表明,ATM-CHK2 轴介导的 ATAD3A 磷酸化 正是该过程的关键环节。这也解释了为何在氧化应激下,ATAD3A 无法继续参与 PINK1 的转运,从而启动自噬。
CHK2调控OPTN磷酸化促进受损线粒体清除
将被泛素化的线粒体靶向至自噬体是线粒体自噬的关键步骤。该过程依赖选择性自噬受体的参与,包括 Optineurin(OPTN)、NDP52、TAX1BP1、p62 和 NBR1。这些受体通过其泛素结合结构域(UBD)识别泛素化线粒体,并通过 LC3 结合区域(LIR)与 LC3 蛋白相互作用。
以往研究表明,TBK1(TANK-binding kinase 1)可在OPTN(Optineurin)的多个位点(Ser473、Ser513、Ser177)进行磷酸化,从而增强 OPTN 与泛素及 LC3 (Microtubule-associated protein 1 light chain 3) 的结合能力,促进自噬体识别受损线粒体。
与此一致,在最新研究中,研究者发现 CHK2 同样能在 Ser177 和 Ser473 位点磷酸化 OPTN,以促进 CCCP 或 AO 诱导的线粒体损伤和氧化应激进而触发的线粒体自噬。不过,TBK1 与 CHK2 的激活机制不同:
TBK1 的活化依赖 OPTN 的泛素结合结构域,而 CHK2 的激活则由 mtROS 信号驱动。这表明 CHK2 代表了一条独立于 TBK1 的 ROS 感应自噬通路。
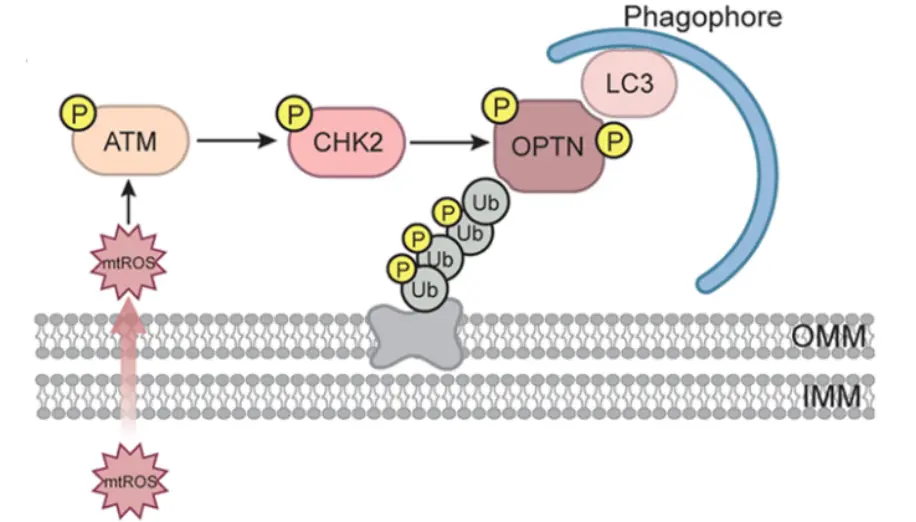
ATM-CHK2-OPTN 轴通过 mtROS 依赖机制驱动泛素化线粒体向自噬体的靶向运输示意图
此外,研究者还发现细胞自噬调控蛋白 Beclin 1 通过启动 自噬体膜形成 促进受损线粒体清除。
当线粒体受到损伤并产生过量的线粒体活性氧(mtROS)时,CHK2 被活化(Thr68位点磷酸化),并与 Beclin 1 的相互作用显著增强。随后,CHK2 直接在 Beclin 1 的 Ser90/Ser93 位点进行磷酸化,促进其从 Bcl-2 复合物中解离,从而解除对自噬的抑制。
实验进一步表明,Beclin 1 的磷酸化状态直接决定自噬体膜的形成能力:野生型 Beclin 1 或模拟磷酸化突变体(Beclin 1-DD)可显著增强 LC3-I 向 LC3-II 的转化并促进自噬体形成,而去磷酸化突变体(Beclin 1-AA)则无法诱导该过程。此外,表达 Beclin 1-WT 或 DD 突变体的细胞显示出更强的线粒体自噬活性,而 AA 突变体细胞自噬明显受抑。
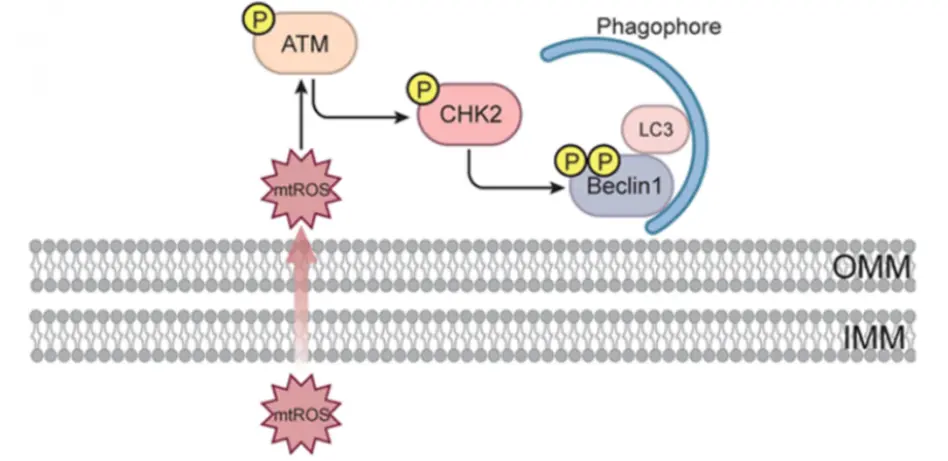
ATM-CHK2-Beclin 1 轴通过感知 mtROS 驱动自噬体膜的形成示意图
氧化应激相关的肾缺血再灌注模型也验证了这一结论。实验中,WT 小鼠表现出 PINK1 积累增加,以及 ATAD3A Ser371、OPTN Ser177/473、Beclin 1 Ser90/93 磷酸化水平升高,线粒体自噬增强,肾损伤减轻,而 Chk2⁻/⁻ 小鼠则未见此变化。这表明 CHK2 介导的线粒体自噬途径在该情况下可促进细胞存活。
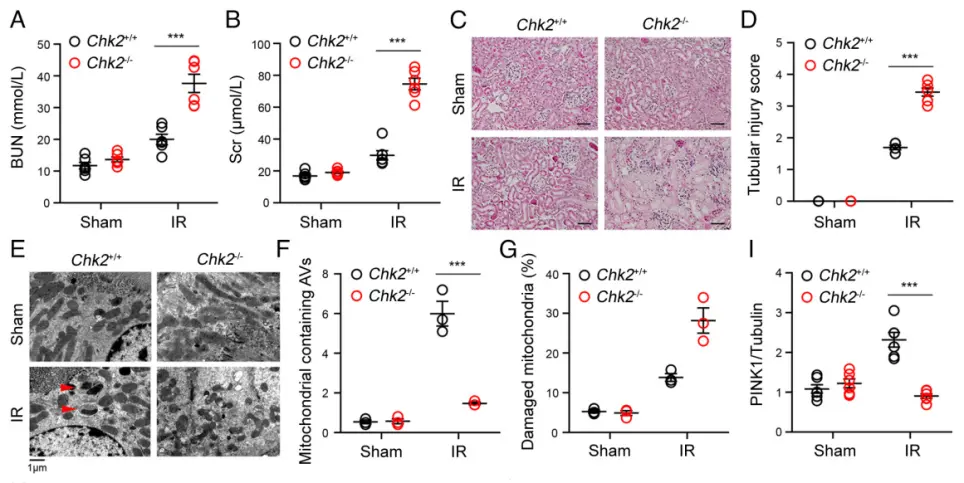
CHK2 介导的线粒体自噬在肾缺血后的组织损伤保护中发挥作用
小结
该研究揭示了线粒体活性氧(mtROS)在启动线粒体自噬(mitophagy)中的核心信号作用,并明确了其分子机制。
研究表明,mtROS 通过激活 DNA 损伤应答通路 ATM-CHK2,促使 CHK2 磷酸化 ATAD3A(Ser371),阻断 PINK1 向内膜转运,导致 PINK1 在外膜积累,从而启动 PINK1/Parkin 依赖的自噬。同时,CHK2 还通过磷酸化自噬受体 OPTN(Ser177/Ser473)增强受损线粒体向自噬体的靶向,并通过 Beclin 1 调控自噬体膜的形成。
体内模型验证显示,CHK2介导的线粒体自噬可减轻缺血再灌注引起的肾损伤,提示该通路在应激条件下有助于维持细胞稳态和存活。
综上,研究明确了 mtROS → ATM-CHK2 → ATAD3A/OPTN/Beclin 1 → PINK1/Parkin 线粒体自噬的调控链,为理解线粒体应激响应和自噬调控机制提供了新的分子基础,同时为潜在的治疗策略提供了理论依据。
科研助力
线粒体是大多数真核细胞中存在的一种离散的细胞器,控制细胞代谢的基本速率,被称为“细胞的发电厂”。然而,线粒体的功能不限于提供细胞能量,线粒体是活性氧物质(ROS)的主要来源。ROS 反映了细胞氧化应激的水平,在细胞凋亡、增殖、衰老等过程发挥重要的信号传导作用。
此外,线粒体是细胞钙稳态的积极参与者。线粒体Ca2+积累调节多种功能,如有氧代谢和诱导细胞死亡。线粒体DNA(mtDNA)的突变是许多线粒体代谢紊乱的原因,并且被认为通过促进细胞凋亡促进衰老。因此线粒体是药物治疗代谢性、退行性(神经退行性疾病)和过度增殖性疾病(癌症)的非常有吸引力的靶点。临床前和临床数据已经证明线粒体靶向方法具有相当大的潜力,小分子药物或生物制剂可通过各种途径作用于线粒体,包括ETC 抑制、OXPHOS 解偶联、线粒体Ca2+调节和ROS 控制等。
TargetMol 线粒体靶向库 收集了950 种线粒体靶向相关的化合物,适合于线粒体靶向药物的研发和相关靶点研究。
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询
版权所有©2015-2026 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4 | 沪公网安备 31010602006700号 | 沪(静)应急管危经许[2024]203441
| 沪(静)应急管危经许[2024]203441