Adenosine 5’-methylenediphosphate is an inhibitor of ecto-5’-nucleotidase, also known as CD73, with a Kivalue of 37 nM.1It inhibits cAMPaccumulation induced by adenosine 5’-monophosphate , adenosine 5’-diphosphate , or adenosine 5’-triphosphate but not adenosine in VA-13 human fibroblasts when used at a concentration of 100 μM. Adenosine 5’-methylenediphosphate reduces proliferation of U138MG glioma cells, as well as inhibits the invasion and migration of MHCC97H hepatocellular carcinoma (HCC) cells in a migration assay.2,3It increases tumor infiltration of CD3+CD8+T cells and reduces tumor growth in a K1735 murine melanoma model when administered at a dose of 400 μg mouse.4 1.Bruns, R.F.Adenosine receptor activation by adenine nucleotides requires conversion of the nucleotides to adenosineNaunyn Schmiedebergs Arch. Pharmacol.315(1)5-13(1980) 2.Braganhol, E., Tamajusuku, A.S.K., Bernardi, A., et al.Ecto-5′-nucleotidase CD73 inhibition by quercetin in the human U138MG glioma cell lineBiochim. Biophys. Acta1770(9)1352-1359(2007) 3.Shali, S., Yu, J., Zhang, X., et al.Ecto 5′ nucleotidase (CD73) is a potential target of hepatocellular carcinomaJ. Cell Physiol.234(7)10248-10259(2018) 4.Forte, G., Sorrentino, R., Montinaro, A., et al.Inhibition of CD73 improves B cell-mediated anti-tumor immunity in a mouse model of melanomaJ. Immunol.189(5)2226-2233(2021)
Peptide YY (PYY) is a 36-amino acid peptide and anorectic gut hormone agonist for the neuropeptide Y receptors Y1, Y2, Y5, and Y6 with EC50 values of 0.7, 0.58, 1, and 0.8 nM, respectively, for supression of forskolin-induced cAMPaccumulation. PYY is cleaved in vivo to PYY (3-36) . Release of PYY occurs postprandially in proportion to calorie intake from intestinal enteroendocrine L cells, indicating it may be a satiety signal. In humans, PYY inhibits food intake and gastrointestinal transit in both lean and obese subjects.
Pituitary adenylate cyclase-activating peptide (PACAP) (6-27) is a PACAP receptor antagonist with IC50 values of 1,500, 600, and 300 nM, respectively, for rat PAC1, rat VPAC1, and human VPAC2 recombinant receptors expressed in CHO cells. It binds to PACAP receptors on SH-SY5Y and SK-N-MC human neuroblastoma and T47D human breast cancer cells (IC50s = 24.5, 106, and 105 nM, respectively) and inhibits cAMPaccumulation induced by PACAP (1-38) (Kis = 457, 102, and 283 nM, respectively, in SH-SY5Y, SK-N-MC, and T47D cells). In vivo, in newborn pigs, PACAP (6-27) (10 μM) inhibits vasodilation of pial arterioles induced by PACAP (1-27) and PACAP (1-38) . It also inhibits PACAP (1-27)-stimulated increases in plasma insulin and glucagon levels and pancreatic venous blood flow in dogs when administered locally to the pancreas at a dose of 500 μg.
Tyr-α-CGRP is an N-terminal extended tyrosinated analogue of α-calcitonin gene-related peptide . It binds to amylin receptors AMY1 and AMY3 in COS-7 cells expressing the human receptors (IC50s = 141 and 1.86 nM, respectively). Tyr-α-CGRP also binds to and stimulates cAMPaccumulation in rat L6 myocytes (IC50 = 4 nM; EC50 = 12 nM). It also binds to rat brain and spleen membrane preparations (IC50s = 0.2 and 0.5 nM, respectively), induces positive chronotropic and inotropic effects in isolated right and left guinea pig atria (EC50s = 282 and 74 nM, respectively), and inhibits the twitch response in rat vas deferens (EC50 = 1.9 nM).
Selective medium-chain free fatty acid receptor GPR84 agonist (EC50 = 139 nM). Exhibits no response at GPR40, GPR41, GPR119 or GPR120 at 100 μM. Activates calcium mobilization, inhibits cAMPaccumulation, and induces ERK1/2 phosphorylation, receptor desensitization and internalization in vitro. Liu et al (2016) Design and synthesis of 2-alkylpyrimidine-4,6-diol and 6-alkylpyridine-2,4-diol as potent GPR84 agonists. ACS Med.Chem.Lett. 7 579 PMID:27326330 |Zhang et al (2016) Discovery and characterization of a novel small-molecule agonist for medium-chain free fatty acid receptor G protein-coupled receptor 84. J.Pharmacol.Exp.Ther. 357 337 PMID:26962172
Bilaid C is a tetrapeptide μ-opioid receptor agonist (Ki= 210 nM in HEK293 cell membranes expressing the human receptor) that has been found inPenicillium.1It inhibits forskolin-induced cAMPaccumulation by 77% in HEK293 cells expressing the human μ-opioid receptor when used at a concentration of 10 μM. Bilaid C induces inward rectifying potassium channel (Kir) currents in rat locus coeruleus slices that endogenously express high levels of the μ-opioid receptor (EC50= 4.2 μM). 1.Dekan, Z., Sianati, S., Yousuf, A., et al.A tetrapeptide class of biased analgesics from an Australian fungus targets the μ-opioid receptorProc. Natl. Acad. Sci. USA116(44)22353-22358(2019)
CAY10680 is a dopamine-sparing, benzothiazinone compound that selectively inhibits both MAO-B activity (IC50 = 34.9 nM in human) and adenosine A2A receptors (Ki = 39.5 nM in human). It demonstrates significantly less potent inhibitory values for other adenosine receptor subtypes (Kis > 1 μM) and MAO-A (IC50 ≥ 10 μM). At 1-20 μM, CAY10680 has been shown to abolish cAMPaccumulation in CHO cells transfected with adenosine A2A receptors. In the central nervous system adenosine A2A receptor expression is localized to dopamine-innervated areas where heteromeric complexes are formed with dopamine D2 receptors. Because inhibition of adenosine A2A receptors has been shown to enhance D2 receptor function, the blockade of adenosine A2A receptors has emerged as a potential treatment for Parkinson's disease. An additional strategy in the treatment of Parkinson's disease has been to block the activity of monoamine oxidase B (MAO-B), the enzyme involved in dopamine catabolism.
Calcitonin is a peptide hormone that lowers blood calcium level and inhibits bone resorption. It belongs to the calcitonin family of peptides, which also includes amylin , calcitonin gene-related peptide , and adrenomedullin. The binding of salmon calcitonin to the human calcitonin receptor (CTR) is not modulated by receptor activity-modifying proteins (RAMPs), which influence affinity of human calcitonin to CTR. Salmon calcitonin binds to human CTR2 with IC50 values of 0.933, 0.224, 0.134, and 0.317 nM alone and with RAMP1, 2, or 3, respectively. It induces cAMPaccumulation in COS-7 cells transfected with CTR2 (EC50 = 0.166 nM). Salmon calcitonin inhibits bone resorption by osteoclasts in a pit formation assay using rat bone slices (ID50 = 0.003 pg mL) and lowers calcium level in vivo in a bioassay of hypocalcemia in rats (ED15 = 33.9 mg kg). Formulations containing salmon calcitonin have been used to treat hypercalcemia, bone destruction by osteoporosis, and Paget's disease.
PSB-17365 is a potent GPR84 agonist. PSB-17365 exhibits EC50 values vs. GPR84 of 2.5nM in a cAMPaccumulation assay, and 100nM in a β-arrestin 2 recruitment assay. No direct binding affinities are provided. PSB-17365 is selective for GPR84 compared to other free fatty acid receptors (FFAR1 and FFAR4). GPR84, a Gi protein-coupled receptor that is activated by medium-chain (hydroxy)fatty acids, appears to play an important role in inflammation, immunity, and cancer.
Tasimelteon-d5 is intended for use as an internal standard for the quantification of tasimelteon by GC- or LC-MS. Tasimelteon is a melatonin (MT) receptor agonist. It selectively binds MT1 and MT2 receptors over a panel of 160 additional receptors and enzymes at 10 µM. Tasimelteon inhibits forskolin-induced cAMPaccumulation with EC50 values of 0.79 and 1 nM in NIH3T3 cells expressing the MT1 or MT2 receptor, respectively. Formulations containing tasimelteon have been used in the treatment of non-24-hour sleep-wake disorder.


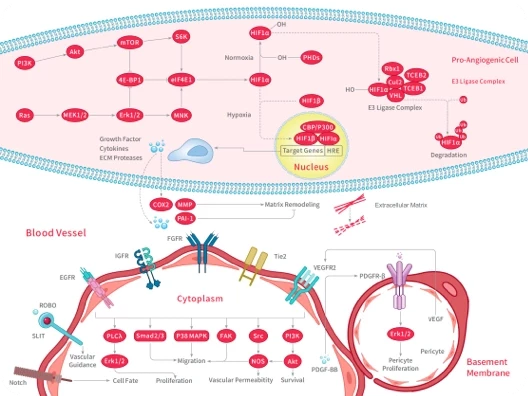
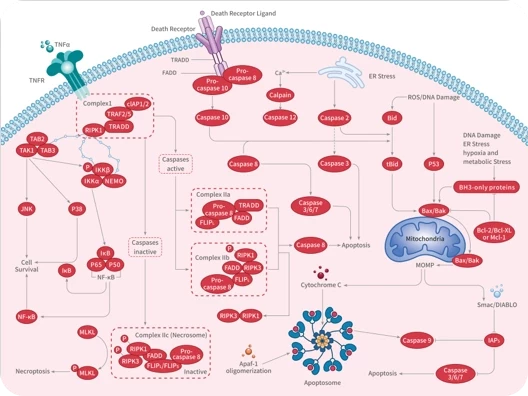
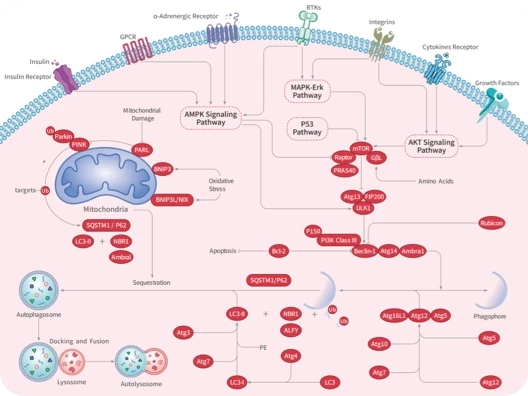

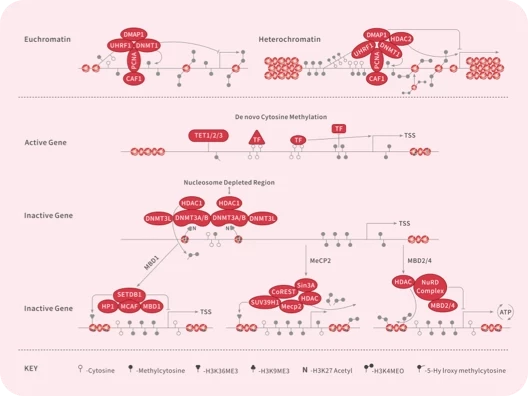

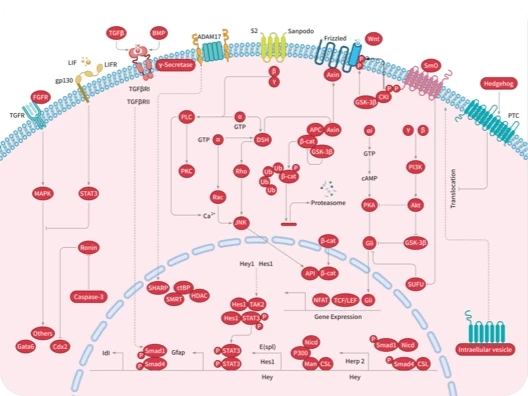
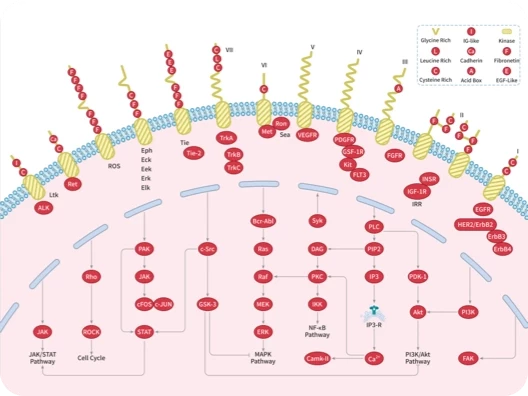
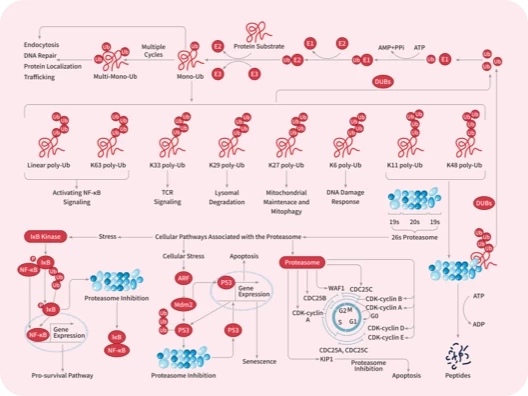
 您的购物车当前为空
您的购物车当前为空












 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询