Potent, non-selective galanin receptor antagonist (Ki values are 1.82 and 5.1 nM at GAL1 and GAL2 respectively) that inhibits galanin (1-29) binding in rat brain in vitro (IC50 = 3 - 15 nM). Attenuates the antidepressant effects of fluoxetine and blocks galanin-induced food intake in vivo. Also exhibits weak partial agonist activity at peripheral GAL2 receptors at doses > 100 nM.
MM-401 is a specific inhibitor of histone H3K4 methyltransferase MLL1 activity that acts by reprogramming mouse epiblast stem cells to naive pluripotency.
Pidnarulex HCl is the salt form of CX-5461, a first-in-class non-genotoxic small molecule targeted inhibitor of RNA polymerase I (Pol I) that activates the p53 pathway without causing DNA damage. CX-5461 selectively inhibits rRNA synthesis by Pol I in the nucleolus, but does not inhibit mRNA synthesis by RNA Polymerase II (Pol II) and does not inhibit DNA replication or protein synthesis. Inhibition of Pol I results in nucleolar stress and release of ribosomal proteins (RP) from the nucleolus. The RP bind to Mdm2 and liberate p53 to orchestrate apoptosis in cancer cells. CX-5461 demonstrates a favorable preclinical profile, potently and selectively kills cancer cells, demonstrates robust in vivo efficacy in multiple models, and has demonstrated oral bioavailability in multiple species.
β-Defensin-2 is a peptide with antimicrobial properties that protects the skin and mucosal membranes of the respiratory, genitourinary, and gastrointestinal tracts.1It inhibits the growth of periodontopathogenic and cariogenic bacteria, includingP. gingivalisandS. salivarius.2β-Defensin-2 (30 μg/ml) stimulates gene expression and production of IL-6, IL-10, CXCL10, CCL2, MIP-3α, and RANTES by keratinocytes.3It also stimulates calcium mobilization, migration, and proliferation of keratinocytes when used at concentrations of 30, 10, and 40 μg/ml, respectively. β-Defensin-2 induces IL-31 production by human peripheral blood-derived mast cellsin vitrowhen used at a concentration of 10 μg/ml and by rat mast cellsin vivofollowing a 500 ng intradermal dose.4Expression of β-defensin-2 is increased in psoriatic skin and chronic wounds.5,6 1.Lehrer, R.I.Primate defensinsNat. Rev. Microbiol.2(9)727-738(2004) 2.Ouhara, K., Komatsuzawa, H., Yamada, S., et al.Susceptibilities of periodontopathogenic and cariogenic bacteria to antibacterial peptides, β-defensins and LL37, produced by human epithelial cellsJ. Antimicrob. Chemother.55(6)888-896(2005) 3.Niyonsaba, F., Ushio, H., Nakano, N., et al.Antimicrobial peptides human β-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokinesJ. Invest. Dermatol.127(3)594-604(2007) 4.Niyonsaba, F., Ushio, H., Hara, M., et al.Antimicrobial peptides human β-defensins and cathelicidin LL-37 induce the secretion of a pruritogenic cytokine IL-31 by human mast cellsJ. Immunol.184(7)3526-3534(2010) 5.Huh, W.-K., Oono, T., Shirafuji, Y., et al.Dynamic alteration of human β-defensin 2 localization from cytoplasm to intercellular space in psoriatic skinJ. Mol. Med. (Berl.)80(10)678-684(2002) 6.Butmarc, J., Yufit, T., Carson, P., et al.Human β-defensin-2 expression is increased in chronic woundsWound Repair Regen.12(4)439-443(2004)
(±)10-HDHA is an autoxidation product of docosahexaenoic acid (DHA) in vitro.[1][2] It is also produced from incubations of DHA in rat liver, brain, and intestinal microsomes.[3][4][5] (±)10-HDHA is a potential marker of oxidative stress in brain and retina where DHA is an abundant polyunsaturated fatty acid. Reference:[1]. VanRollins, M., and Murphy, R.C. Autooxidation of docosahexaenoic acid: Analysis of ten isomers of hydroxydocosahexaenoate. J. Lipid Res. 25(5), 507-517 (1984).[2]. Reynaud, D., Thickitt, C.P., and Pace-Asciak, C.R. Facile preparation and structural determination of monohydroxy derivatives of docosahexaenoic acid (HDoHE) by α-tocopherol-directed autoxidation. Anal. Biochem. 214(1), 165-170 (1993).[3]. VanRollins, M., Baker, R.C., Sprecher, H., et al. Oxidation of docosahexaenoic acid by rat liver microsomes. J. Biol. Chem. 259(9), 5776-5783 (1984).[4]. Yamane, M., Abe, A., and Yamane, S. High-performance liquid chromatography-thermospray mass spectrometry of epoxy polyunsaturated fatty acids and epoxyhydroxy polyunsaturated fatty acids from an incubation mixture of rat tissue homogenate. J. Chromatogr. 652(2), 123-136 (1994).[5]. Kim, H.Y., Karanian, J.W., Shingu, T., et al. Sterochemical analysis of hydroxylated docosahexaenoates produced by human platelets and rat brain homogenate. Prostaglandins 40(5), 473-490 (1990).
Urocortin III is a neuropeptide hormone and member of the corticotropin-releasing factor (CRF) family which includes mammalian CRF , urocortin , urocortin II , frog sauvagine, and piscine urotensin I.1 Human urocortin III shares 90, 40, 37, and 21% identity to mouse urocortin III , mouse urocortin II , human urocortin , and mouse urocortin, respectively. Urocortin III selectively binds to type 2 CRF receptors (Kis = 21.7, 13.5, and >100 nM for rat CRF2α, rat CRF2β, and human CRF1, respectively). It stimulates cAMP production in CHO cells expressing rat CRF2α and mouse CRF2β (EC50s = 0.16 and 0.12 nM, respectively) as well as cultured anterior pituitary cells expressing endogenous CRF2β. Urocortin III is co-released with insulin to potentiate glucose-stimulated somatostatin release in vitro in human pancreatic β-cells.2 In vivo, urocortin III reduces food intake in a dose- and time-dependent manner in mice with a minimum effective dose (MED) of 0.3 nmol/animal.3 It increases swimming time in a forced swim test in mice, indicating antidepressant-like activity.4References1. Lewis, K., Li, C., Perrin, M.H., et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc. Natl. Acad. Sci. U.S.A. 98(13), 7570-7575 (2001).2. van der Meulen, T., Donaldson, C.J., Cáceres, E., et al. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 21(7), 769-776 (2015).3. Pelleymounter, M.A., Joppa, M., Ling, N., et al. Behavioral and neuroendocrine effects of the selective CRF2 receptor agonists urocortin II and urocortin III. Peptides 25(4), 659-666 (2004).4. Tanaka, M., Kádár, K., Tóth, G., et al. Antidepressant-like effects of urocortin 3 fragments. Brain Res. Bull. 84(6), 414-418 (2011). Urocortin III is a neuropeptide hormone and member of the corticotropin-releasing factor (CRF) family which includes mammalian CRF , urocortin , urocortin II , frog sauvagine, and piscine urotensin I.1 Human urocortin III shares 90, 40, 37, and 21% identity to mouse urocortin III , mouse urocortin II , human urocortin , and mouse urocortin, respectively. Urocortin III selectively binds to type 2 CRF receptors (Kis = 21.7, 13.5, and >100 nM for rat CRF2α, rat CRF2β, and human CRF1, respectively). It stimulates cAMP production in CHO cells expressing rat CRF2α and mouse CRF2β (EC50s = 0.16 and 0.12 nM, respectively) as well as cultured anterior pituitary cells expressing endogenous CRF2β. Urocortin III is co-released with insulin to potentiate glucose-stimulated somatostatin release in vitro in human pancreatic β-cells.2 In vivo, urocortin III reduces food intake in a dose- and time-dependent manner in mice with a minimum effective dose (MED) of 0.3 nmol/animal.3 It increases swimming time in a forced swim test in mice, indicating antidepressant-like activity.4 References1. Lewis, K., Li, C., Perrin, M.H., et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc. Natl. Acad. Sci. U.S.A. 98(13), 7570-7575 (2001).2. van der Meulen, T., Donaldson, C.J., Cáceres, E., et al. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 21(7), 769-776 (2015).3. Pelleymounter, M.A., Joppa, M., Ling, N., et al. Behavioral and neuroendocrine effects of the selective CRF2 receptor agonists urocortin II and urocortin III. Peptides 25(4), 659-666 (2004).4. Tanaka, M., Kádár, K., Tóth, G., et al. Antidepressant-like effects of urocortin 3 fragments. Brain Res. Bull. 84(6), 414-418 (2011).
PKI-179 is a potent and orally active dual PI3K mTOR inhibitor, with IC50s of 8 nM, 24 nM, 74 nM, 77 nM, and 0.42 nM for PI3K-α, PI3K-β, PI3K-γ, PI3K-δ and mTOR, respectively. PKI-179 also exhibits activity over E545K and H1047R, with IC50s of 14 nM and 11 nM, respectively. PKI-179 shows anti-tumor activity in vivo[1][2]. PKI-179 inhibits the cell proliferation, with IC50s of 22 nM and 29 nM for MDA361 and PC3 cells, respectively[1].PKI-179 shows inhibitory activity against a panel of 361 other kinases, hERG and cytochrome P450 (CYP) isoforms at concentrations up to >30 μM, but does have activity for CYP2C8 (IC50=3 μM)[1]. PKI-179 (5-50 mg kg; p.o. once daily for 40 days) inhibits the tumor growth and is well tolerated in nude mice bearing MDA-361 human breast cancer tumors[1].PKI-179 (50 mg kg; p.o.) results in good inhibition of PI3K signaling in nude mice bearing MDA361 tumor xenografts[1].PKI-179 exhibits good oral bioavailability (98% in nude mouse, 46% in rat, 38% in monkey, and 61% in dog) and a high half-life (>60 min) [1]. [1]. Venkatesan AM, et, al. PKI-179: an orally efficacious dual phosphatidylinositol-3-kinase (PI3K) mammalian target of rapamycin (mTOR) inhibitor. Bioorg Med Chem Lett. 2010 Oct 1;20(19):5869-73.[2]. Rehan M. A structural insight into the inhibitory mechanism of an orally active PI3K mTOR dual inhibitor, PKI-179 using computational approaches. J Mol Graph Model. 2015 Nov;62:226-234.
Alaproclate is a selective serotonin reuptake inhibitor (SSRI).1,2 It inhibits depletion of serotonin (5-HT) induced by 4-methyl-α-ethyl-m-tyramine in rat cerebral cortex, hippocampus, hypothalamus, and striatum (EC50s = 18, 4, 8, and 12 mg kg, respectively).1 Alaproclate inhibits NMDA-evoked currents and depolarization-induced voltage-dependent potassium currents in rat hippocampal neurons (IC50s = 1.1 and 6.9 μM, respectively) and does not inhibit GABA-evoked currents when used at concentrations up to 100 μM.2 It increases sirtuin 1 (SIRT1) levels in N2a murine neuroblastoma cells expressing apolipoprotein E4 (ApoE4; IC50 = 2.3 μM) and in the hippocampus in the FXFAD-ApoE4 transgenic mouse model of Alzheimer's disease when administered at a dose of 20 mg kg twice daily.3 Alaproclate (40 mg kg) decreases immobility time in the forced swim test in rats, indicating antidepressant-like activity.4References1. Michael, G.B., Eidam, C., Kadlec, K., et al. Increased MICs of gamithromycin and tildipirosin in the presence of the genes erm(42) and msr(E)-mph(E) for bovine Pasteurella multocida and Mannheimia haemolytica. Journal of Antimicrobial Chemotherapy 67(6), 1555-1557 (2012).2. Svensson, B.E., Werkman, T.R., and Rogawski, M.A. Alaproclate effects on voltage-dependent K+ channels and NMDA receptors: Studies in cultured rat hippocampal neurons and fibroblast cells transformed with Kv1.2 K+ channel cDNA. Neuropharmacology 33(6), 795-804 (1994).3. Campagna, J., Soilman, P., Jagodzinska, B., et al. A small molecule ApoE4-targeted therapeutic candidate that normalizes sirtuin 1 levels and improves cognition in an Alzheimer's disease mouse model. Sci. Rep. 8(1), 17574 (2018).4. Danysz, W.P., A., Kostowski, W., Malatynska, E., et al. Comparison of desipramine, amitriptyline, zimeldine and alaproclate in six animal models used to investigate antidepressant drugs. Pharmacol. Toxicol. 62(1), 42-50 (1988). Alaproclate is a selective serotonin reuptake inhibitor (SSRI).1,2 It inhibits depletion of serotonin (5-HT) induced by 4-methyl-α-ethyl-m-tyramine in rat cerebral cortex, hippocampus, hypothalamus, and striatum (EC50s = 18, 4, 8, and 12 mg kg, respectively).1 Alaproclate inhibits NMDA-evoked currents and depolarization-induced voltage-dependent potassium currents in rat hippocampal neurons (IC50s = 1.1 and 6.9 μM, respectively) and does not inhibit GABA-evoked currents when used at concentrations up to 100 μM.2 It increases sirtuin 1 (SIRT1) levels in N2a murine neuroblastoma cells expressing apolipoprotein E4 (ApoE4; IC50 = 2.3 μM) and in the hippocampus in the FXFAD-ApoE4 transgenic mouse model of Alzheimer's disease when administered at a dose of 20 mg kg twice daily.3 Alaproclate (40 mg kg) decreases immobility time in the forced swim test in rats, indicating antidepressant-like activity.4 References1. Michael, G.B., Eidam, C., Kadlec, K., et al. Increased MICs of gamithromycin and tildipirosin in the presence of the genes erm(42) and msr(E)-mph(E) for bovine Pasteurella multocida and Mannheimia haemolytica. Journal of Antimicrobial Chemotherapy 67(6), 1555-1557 (2012).2. Svensson, B.E., Werkman, T.R., and Rogawski, M.A. Alaproclate effects on voltage-dependent K+ channels and NMDA receptors: Studies in cultured rat hippocampal neurons and fibroblast cells transformed with Kv1.2 K+ channel cDNA. Neuropharmacology 33(6), 795-804 (1994).3. Campagna, J., Soilman, P., Jagodzinska, B., et al. A small molecule ApoE4-targeted therapeutic candidate that normalizes sirtuin 1 levels and improves cognition in an Alzheimer's disease mouse model. Sci. Rep. 8(1), 17574 (2018).4. Danysz, W.P., A., Kostowski, W., Malatynska, E., et al. Comparison of desipramine, amitriptyline, zimeldine and alaproclate in six animal models used to investigate antidepressant drugs. Pharmacol. Toxicol. 62(1), 42-50 (1988).
TAS-103 is a dual inhibitor of DNA topoisomerase I II, used for cancer research. TAS-103 is a dual inhibitor of DNA topoisomerase I II. TAS-103 (0.1-10 μM) is active on CCRF-CEM cells, with an IC50 value of 5 nM. TAS-103 (0.1 μM) significantly increases levels of topo IIα FITC immunofluorescence in individual CCRF-CEM cells[1]. TAS-103 (0.01-1 μM) is highly cytotoxic to Lewis lung carcinoma (LLC) cells, and Liposomal TAS-103 is almost as active as free TAS-103[2]. TAS-103 inhibits the viability of HeLa cells, with an IC50 of 40 nM. TAS-103 (10 μM) disrupts signal recognition particle (SRP) complex formation, and induces destabilization of SRP14 and SRP19 and its eventual degradation[3]. TAS-103 (30 mg kg, i.v.) causes significant tumor growth suppression in mice bearing Lewis lung carcinoma (LLC) cells, without obvious body weight loss, and the liposomal TAS-103 is more active than free TAS-103[2]. [1]. Padget K, et al. An investigation into the formation of N- [2-(dimethylamino)ethyl]acridine-4-carboxamide (DACA) and 6-[2-(dimethylamino)ethylamino]- 3-hydroxy-7H-indeno[2, 1-C]quinolin-7-one dihydrochloride (TAS-103) stabilised DNA topoisomerase I and II cleavable complexes in human leukaemia cells. Biochem Pharmacol. 2000 Sep 15;60(6):817-21. [2]. Shimizu K, et al. Cancer chemotherapy by liposomal 6-[12-(dimethylamino)ethyl]aminol-3-hydroxy-7H-indeno[2,1-clquinolin-7-one dihydrochloride (TAS-103), a novel anti-cancer agent. Biol Pharm Bull. 2002 Oct;25(10):1385-7. [3]. Yoshida M, et al. A new mechanism of 6-((2-(dimethylamino)ethyl)amino)-3-hydroxy-7H-indeno(2,1-c)quinolin-7-one dihydrochloride (TAS-103) action discovered by target screening with drug-immobilized affinity beads. Mol Pharmacol. 2008 Mar;73(3):987-94. Epub 2007 Dec 18.
9(S),12(S),13(S)-TriHOME is a linoleic acid-derived oxylipin that has diverse biological activities.1,2,3,4It has been found in various plants and is produced in human eosinophils in a 15-lipoxygenase-dependent, soluble epoxide hydrolase-independent manner.1,59(S),12(S)13(S)-TriHOME inhibits antigen-induced β-hexosaminidase release from RBL-2H3 mast cells (IC50= 28.7 μg ml).2It inhibits LPS-induced nitric oxide (NO) production in BV-2 microglia (IC50= 40.95 μM).3In vivo, 9(S),12(S),13(S)-TriHOME (1 g animal) enhances the antiviral IgA and IgG antibody responses induced by a nasal influenza hemagglutinin (HA) vaccine by 5.2- and 2-fold, respectively, in mice.4 1.Hamberg, M., and Hamberg, G.Peroxygenase-catalyzed fatty acid epoxidation in cereal seeds: Sequential oxidation of linoleic acid into 9(S),12(S),13(S)-trihydroxy-10(E)-octadecenoic acidPlant Physiol.110(3)807-815(1996) 2.Hong, S.S., and Oh, J.S.Inhibitors of antigen-induced degranulation of RBL-2H3 cells isolated from wheat branJ. Korean Soc. Appl. Biol. Chem.5569-74(2012) 3.Kim, C.S., Kwon, O.W., Kim, S.Y., et al.Five new oxylipins from Chaenomeles sinensisLipids49(11)1151-1159(2014) 4.Shirahata, T., Sunazuka, T., Yoshida, K., et al.Total synthesis, elucidation of absolute stereochemistry, and adjuvant activity of trihydroxy fatty acidsTetrahedron62(40)9483-9496(2006) 5.Fuchs, D., Tang, X., Johnsson, A.-K., et al.Eosinophils synthesize trihydroxyoctadecenoic acids (TriHOMEs) via a 15-lipoxygenase dependent processBiochim. Biophys. Acta Mol. Cell Biol. Lipids1865(4)158611(2020)
Glycerophosphorylethanolamine is an active phosphodiester metabolite of phosphatidylethanolamine.1,2It promotes aggregation of amyloid-β (1-40) (Aβ40)in vitro, and levels of glycerophosphorylethanolamine are elevated in postmortem brains isolated from patients with Alzheimer’s disease. 1.Klunk, W.E., Xu, C.J., McClure, R.J., et al.Aggregation of β-amyloid peptide is promoted by membrane phospholipid metabolites elevated in Alzheimer’s disease brainJ. Neurochem.69(1)266-272(1997) 2.Blusztajn, J.K., Lopez Gonzalez-Coviella, I., Logue, M., et al.Levels of phospholipid catabolic intermediates, glycerophosphocholine and glycerophosphoethanolamine, are elevated in brains of Alzheimer’s disease but not of Down’s syndrome patientsBrain Res.536(1-2)240-244(1990)
Carbazomycin D is a bacterial metabolite that has been found inStreptomycesand has diverse biological activities.1,2It is active against the fungiT. asteroidesandT. mentagrophytes(MIC = 100 μg ml for both) and the bacteriumM. tuberculosis(IC50= 25 μg ml). Carbazomycin D is cytotoxic to MCF-7, KB, NCI H187, and Vero cells (IC50s = 21.3, 33.2, 12.9, and 34.3 μg ml, respectively).2 1.Naid, T., Kitahara, T., Kaneda, M., et al.Carbazomycins C, D, E and F, minor components of the carbazomycin complexJ. Antibiot. (Tokyo)40(2)157-164(1987) 2.Intaraudom, C., Rachtawee, P., Suvannakad, R., et al.Antimalarial and antituberculosis substances from Streptomyces sp. BCC26924Tetrahedron67(39)7593-7597(2011)



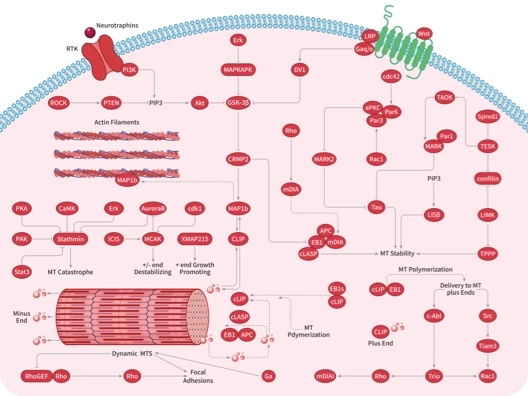
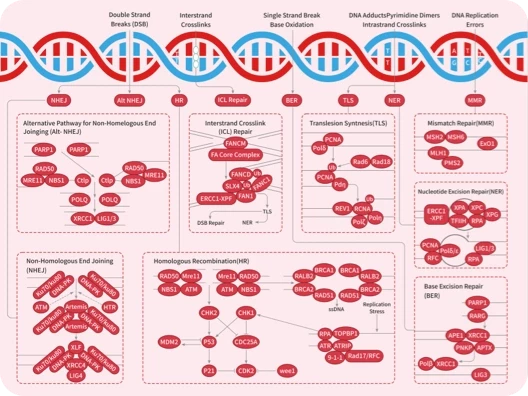
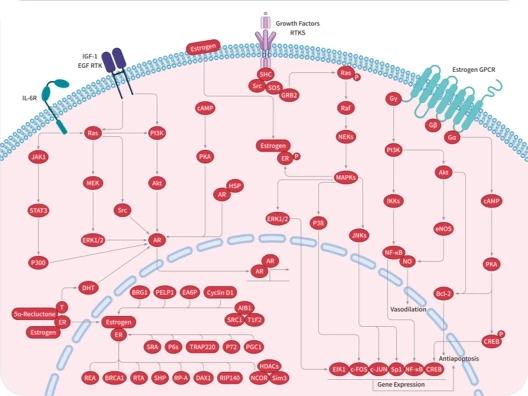
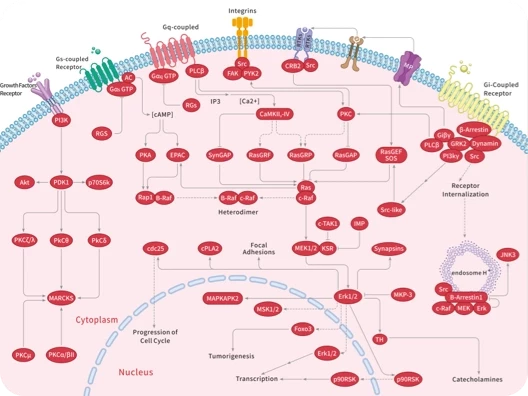
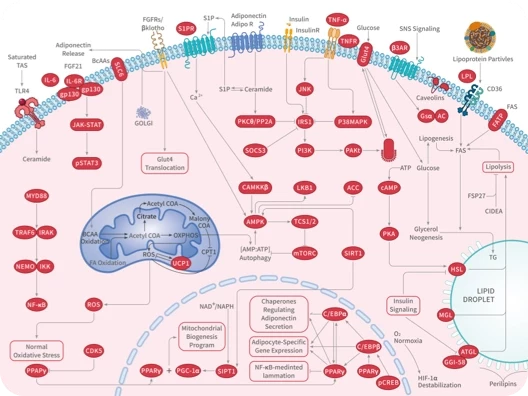
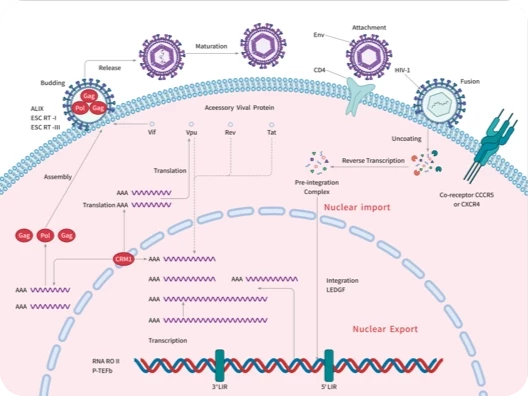

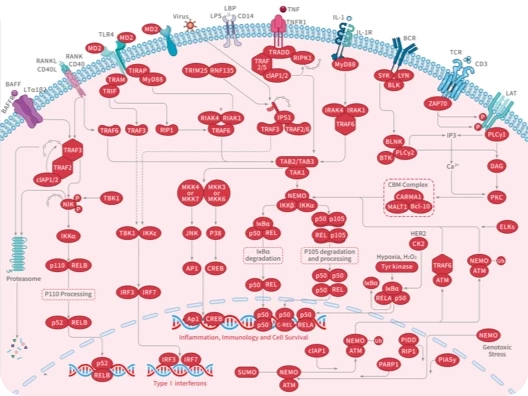
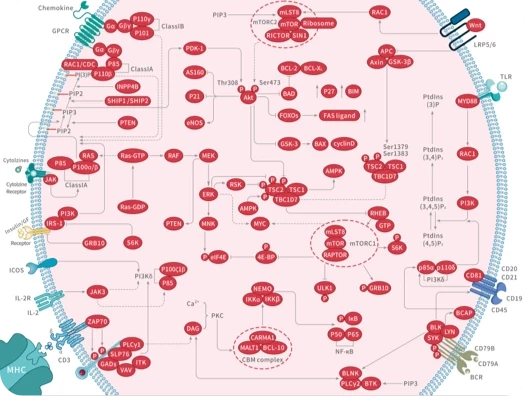
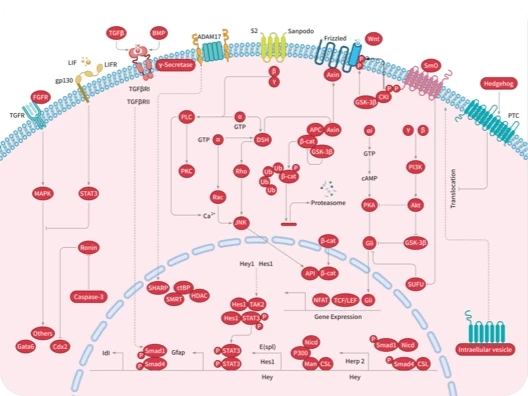
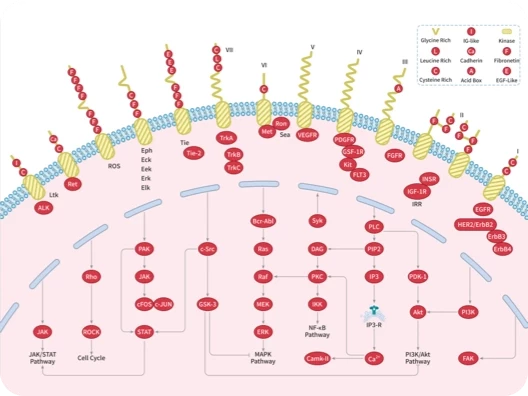
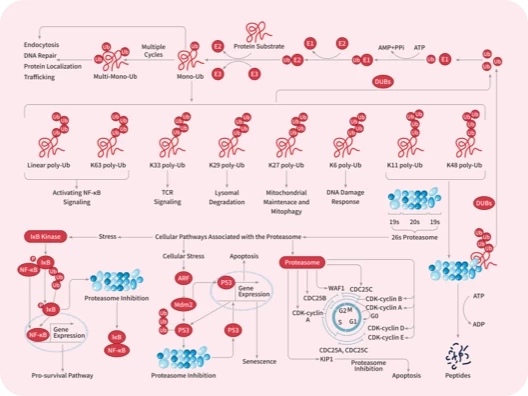
 您的购物车当前为空
您的购物车当前为空












 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询