β-Defensin-3 is a peptide with antimicrobial properties that protects the skin and mucosal membranes of the respiratory, genitourinary, and gastrointestinal tracts. It inhibits the growth of the periodontopathogenic and cariogenic bacteria F. nucleatum, S. mutans, S. sobrinus, S. salivarius, and L. casei (MICs = 12.5-100 mg/l). It also inhibits the growth of S. aureus, S. pyogenes, P. aeruginosa, E. coli, and C. albicans. β-Defensin-3 stimulates gene expression and production of IL-6, IL-10, CXCL10, CCL2, MIP-3α, and RANTES by keratinocytes when used at a concentration of 30 μg/ml. It also stimulates calcium mobilization, migration, and proliferation of keratinocytes when used at concentrations of 30, 5, and 20 μg/ml, respectively. β-Defensin-3 induces IL-31 production by human peripheral blood-derived mastcells in vitro when used at a concentration of 10 μg/ml and by rat mastcells in vivo following a 500 ng intradermal dose.
β-Defensin-4 is a peptide with antimicrobial properties that protects the skin and mucosal membranes of the respiratory, genitourinary, and gastrointestinal tracts. It induces migration of monocytes in vitro when used at a concentration of 10 nM but does not affect migration of neutrophils and eosinophils. β-Defensin-4 (30 μg/ml) stimulates gene expression and production of IL-6, IL-10, CXCL10, CCL2, MIP-3α, and RANTES by keratinocytes. It also stimulates calcium mobilization, migration, and proliferation of keratinocytes when used at concentrations of 30, 10, and 40 μg/ml, respectively. β-Defensin-4 induces IL-31 production by human peripheral blood-derived mastcells in vitro when used at a concentration of 10 μg/ml and by rat mastcells in vivo following a 500 ng intradermal dose. It also inhibits growth of E. coli, P. aeruginosa, and S. aureus with lethal concentration (LC) values of 5, 12, and 15 μM, respectively, of S. carnosus (MIC = 4.5 μg/ml), and of C. albicans with a minimum fungicidal concentration (MFC) value of 7.5 μM.
Deethylindanomycin is a polyether antibiotic that has been found in S. setonii. It is active against a variety of Gram-positive bacteria, including various strains of S. aureus and Streptococcus, as well as one strain of S. pneumoniae (MICs = 4, 4, and 2 μg ml, respectively). It is also active against coccidia in vitro, inhibiting E. tenella development, but is inactive against E. tenella infection in chicks when administered at a dose of 200 μg g in the diet. Deethylindanomycin acts as an ionophore in lipid bilayer membranes and is more selective for potassium ions than calcium, magnesium, and sodium ions. It induces histamine release from rodent mastcells and human basophils in vitro in a calcium-dependent manner.
Prostaglandin D2 (PGD2) is the major eicosanoid product of mastcells and is produced in large quantities by hematopoietic PGD synthase during allergic and asthmatic anaphylaxis. It causes vasodilation, flushing, hypotension, and is an inhibitor of platelet aggregation. Prostaglandin D2 methyl ester (PGD2 methyl ester) is a more lipid-soluble, cell-permeable prodrug form of PGD2. It binds to the human and mouse PGD2 receptors (DP1 and CRTH2 DP2) with 5-10 fold lower affinity than PGD2.
Streptochlorin is a bacterial metabolite originally isolated from Streptomyces sp. SF2583 that has diverse biological activities, including antiangiogenic, antiproliferative, and anti-allergic properties. It inhibits TNF-α-induced NF-κB transcriptional activity and decreases proliferation of human umbilical vein endothelial cells (HUVECs) when used at concentrations ranging from 5 to 20 μM. Streptochlorin (12 μg/ml) decreases viability of, as well as induces apoptosis and increases the production of reactive oxygen species (ROS) in, Hep3B human hepatocellular carcinoma cells. It does not induce cytotoxicity in RBL-2H3 mastcells at concentrations up to 100 μM. Streptochlorin prevents degranulation in antigen-stimulated mastcells, as well as inhibits Syk kinase and the Src family kinases LYN and Fyn and reduces the secretion of TNF-α and IL-4 induced by dinitrophenyl-human serum album (DNP-HSA) in RBL-2H3 mastcells. It also decreases swelling and reduces scratching behavior in a mouse model of allergic dermatitis induced by dinitrofluorobenzene (DNFB).
14,15-Leukotriene D4 (14,15-LTD4) is a member of an alternate class of LTs synthesized by a pathway involving the dual actions of 15- and 12-lipoxygenases (15- and 12-LOs) on arachidonic acid via 15-HpETE and 14,15-LTA4 intermediates. 14,15-LTD4 is classified as an eoxin (EXD4), because it is formed mostly by eosinophils. However, mastcells and nasal polyps can synthesize 14,15-LTD4 as well. Little is known about the physiological actions of 14,15-LTD4. It has weak contractile activity on both guinea pig ileum and pulmonary parenchyma in contrast to the effects of 5-LO-derived LTs. However, in an in vitro permeability assay, 14,15-LTD4 can increase vascular permeability of human endothelial cell monolayers, with similar potency to that of 5-LO-derived LTs, resulting in plasma leakage, a hallmark of inflammation.
Leukotrienes (LTs) are a group of acute inflammatory mediators derived from arachidonic acid in leukocytes. The majority of these metabolites are formed through the 5-lipoxygenase (5-LO) pathway. 14,15-LTE4 is a metabolite of 14,15-LTC4 and 14,15-LTD4, an alternate class of LTs synthesized by a pathway involving the dual actions of 15- and 12-LOs on arachidonic acid via 15-HpETE and 14,15-LTA4 intermediates. These metabolites are classified as eoxins because they are formed mostly by eosinophils. Mastcells and nasal polyps can synthesize 14,15-LTC4 as well, however metabolism to 14,15-LTE4 in these cells and tissue has not been documented. 14,15-LTE4 increases vascular permeability of human endothelial cell monolayers with about 10-fold less potency than LTC4, but approximately 100-fold greater potency than histamine.
9(S),12(S),13(S)-TriHOME is a linoleic acid-derived oxylipin that has diverse biological activities.1,2,3,4It has been found in various plants and is produced in human eosinophils in a 15-lipoxygenase-dependent, soluble epoxide hydrolase-independent manner.1,59(S),12(S)13(S)-TriHOME inhibits antigen-induced β-hexosaminidase release from RBL-2H3 mastcells (IC50= 28.7 μg ml).2It inhibits LPS-induced nitric oxide (NO) production in BV-2 microglia (IC50= 40.95 μM).3In vivo, 9(S),12(S),13(S)-TriHOME (1 g animal) enhances the antiviral IgA and IgG antibody responses induced by a nasal influenza hemagglutinin (HA) vaccine by 5.2- and 2-fold, respectively, in mice.4 1.Hamberg, M., and Hamberg, G.Peroxygenase-catalyzed fatty acid epoxidation in cereal seeds: Sequential oxidation of linoleic acid into 9(S),12(S),13(S)-trihydroxy-10(E)-octadecenoic acidPlant Physiol.110(3)807-815(1996) 2.Hong, S.S., and Oh, J.S.Inhibitors of antigen-induced degranulation of RBL-2H3 cells isolated from wheat branJ. Korean Soc. Appl. Biol. Chem.5569-74(2012) 3.Kim, C.S., Kwon, O.W., Kim, S.Y., et al.Five new oxylipins from Chaenomeles sinensisLipids49(11)1151-1159(2014) 4.Shirahata, T., Sunazuka, T., Yoshida, K., et al.Total synthesis, elucidation of absolute stereochemistry, and adjuvant activity of trihydroxy fatty acidsTetrahedron62(40)9483-9496(2006) 5.Fuchs, D., Tang, X., Johnsson, A.-K., et al.Eosinophils synthesize trihydroxyoctadecenoic acids (TriHOMEs) via a 15-lipoxygenase dependent processBiochim. Biophys. Acta Mol. Cell Biol. Lipids1865(4)158611(2020)
11-trans Leukotriene C4 (11-trans LTC4) is a C-11 double bond isomer of LTC4. LTC4 undergoes slow temperature-dependent isomerization to 11-trans LTC4 during storage. 11-trans LTC4 is produced in smaller amounts relative to LTC4 in ionophore-stimulated HMC-1 cells (a humanmast cell line) and equine eosinophils, but not in human neutrophils or RBL-1 cells. It is nearly equipotent with LTC4 for contraction of guinea pig parenchymal and ileum. In a radioligand binding assay using guinea pig ileum as a cysteinyl leukotriene receptor preparation, the pKis for LTC4 and 11-trans LTC4 were determined to be 6.42 and 6.58, respectively.
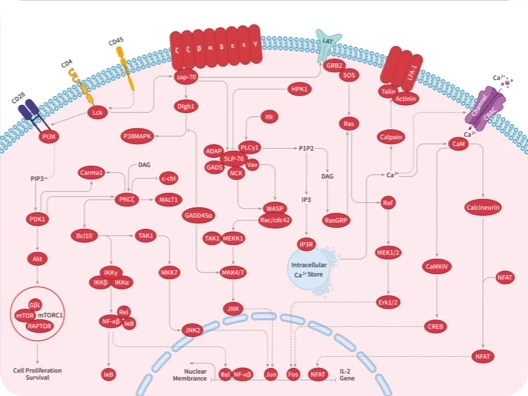
Interleukin-2-inducible T cell kinase (ITK) is a non-receptor tyrosine kinase expressed in T cells, NKT cells and mastcells which plays a crucial role in regulating the T cell receptor (TCR), CD28, CD2, chemokine receptor CXCR4, and FcepsilonR-mediated signaling pathways. ITK inhibitors can be used for the treatment of inflammation and immune-mediated disorders. ITK inhibitor (N-[5-[[3-[(4-Acetylpiperazin-1-yl)carbonyl]-4-methyl-6-methoxy-phenyl]thio]thiazol-2-yl]-4-(N-1,2-dimethylpropylaminomethyl)benzamide) is the analogue of BMS-509744, which can potently and selectively inhibit Itk kinase activity. In vitro: BMS-509744 could reduce TCR-induced functions including PLCγ1 tyrosine phosphorylation, calcium mobilization, IL-2 secretion, and T-cell proliferation in vitro in both human and mouse cells [1]. In vivo: BMS-509744 suppressed the production of IL-2 induced by anti-TCR antibody administered to mice. BMS-509744 also significantly diminishes lung inflammation in a mouse model of ovalbumin-induced allergy/asthma [1]. Clinical trial: Up to now, both BMS-509744 and ITK inhibitor is still in the preclinical development stage.
Produced by neutrophils, macrophages, mastcells, and by transcellular metabolism in platelets, leukotriene C4 (LTC4) is the parent cysteinyl leukotriene formed by the LTC4 synthase-catalyzed conjugation of glutathione to LTA4. It is one of the constituents of slow-reacting substance of anaphylaxis (SRS-A) and exhibits potent smooth muscle contracting activity. LTC4, however, is rapidly metabolized to LTD4 and LTE4, which makes the characterization of LTC4 pharmacology difficult. N-methyl Leukotriene C4 (N-methyl LTC4) is a synthetic analog of LTC4 that is not readily metabolized to LTD4 and LTE4.It acts as a potent and selective CysLT2 receptor agonist exhibiting EC50 values of 122 and > 2,000 nM at the human CysLT2 and CysLT1 receptors, respectively. It has essentially the same potency as LTC4 at both the human and murine receptors CysLT2 receptors. N-methyl LTC4 is potent and active in vivo, causing vascular leak in mice overexpressing the human CysLT2 receptor but not in CysLT2 receptor knockout mice.
Negative control for PMX 53. Active Analog also available. Subramanian et al (2011) PMX-53 as a dual CD88 antagonist and an agonist for Mas-related gene 2 (MrgX2) in humanmastcells. Mol.Pharmacol. 79 1005 PMID:21441599
Prostaglandin D2 is synthesized by hematopoietic-type PGD-synthase (H-PGDS) in mastcells and is released in large quantities during allergic and asthmatic anaphylaxis. PGD2 is also produced in the brain by lipocalin-PGD-synthase also known as β-trace. In the brain, PGD2 produces normal physiological sleep and lowering of body temperature. Further pharmacological actions include inhibition of platelet aggregation and relaxation of vascular smooth muscle. tetranor-PGDM is a major metabolite of PGD2 that is detectable in human and mouse urine. The levels of tetranor-PGDM and 2,3-dinor-11β-PGF2α , a related PGD2 metabolite, in human urine were found to be 1.5 ± 0.3 and 0.6 ± ng mg creatinine, respectively. tetranor-PGDM was detected in murine urine at a level of 8.1 ± 1.3 ng mg creatinine.
Abarelix acetate is a synthetic third generation gonadotropin-releasing hormone receptor (GnRHR) antagonist. It increases histamine release from rat peritoneal mastcells in vitro and from a human skin model ex vivo. In vivo, abarelix decreases plasma luteinizing hormone (LH) levels six hours post-treatment in castrated rats, with levels returning to baseline within 24 hours.3 Abarelix (2 mg kg) also transiently decreases plasma testosterone levels in intact rats, with levels returning to baseline within seven days post-treatment. Formulations containing abarelix have previously been used in the treatment of advanced prostate cancer.
NBI-42902 is a potent inhibitor of peptide radioligand binding to the human GnRH receptor (K(i) = 0.56 nm). Tritiated NBI-42902 binds with high affinity (K(d) = 0.19 nm) to a single class of binding sites and can be displaced by a range of peptide and nonpeptide GnRH receptor ligands. In vitro experiments demonstrate that NBI-42902 is a potent functional, competitive antagonist of GnRH stimulated IP accumulation, Ca(2+) flux, and ERK1 2 activation. It did not stimulate histamine release from rat peritoneal mastcells. Finally, it is effective in lowering serum LH in castrated male macaques after oral administration. Overall, these data provide a benchmark of pharmacological characteristics required for a nonpeptide GnRH antagonist to effectively suppress gonadotropins in humans and suggest that NBI-42902 may have clinical utility as an oral agent for suppression of the hypothalamic-pituitary-gonadal axis. (source: Endocrinology. 2007 Feb;148(2):857-67. Epub 2006 Nov 9.)



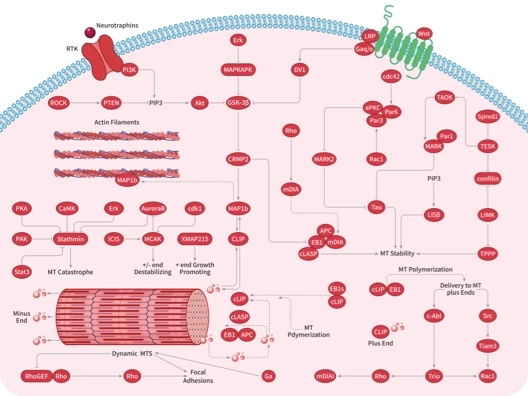
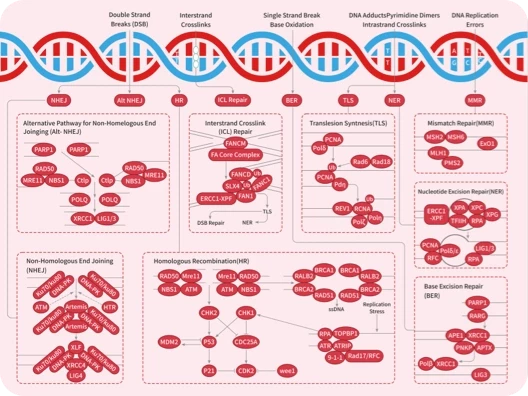
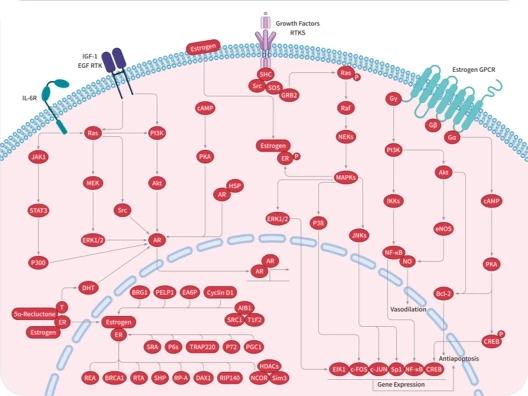
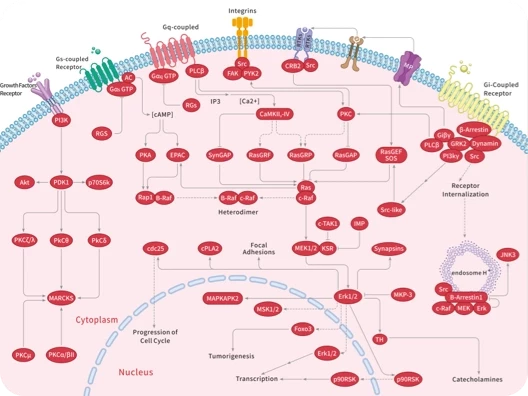

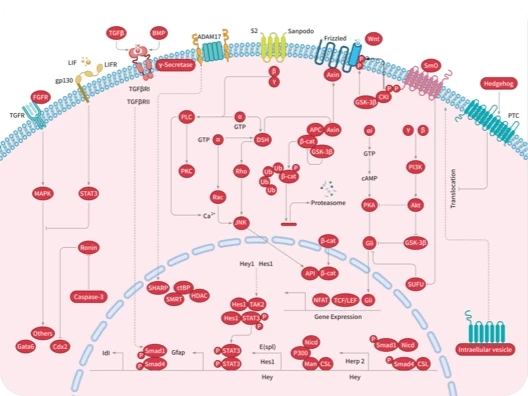
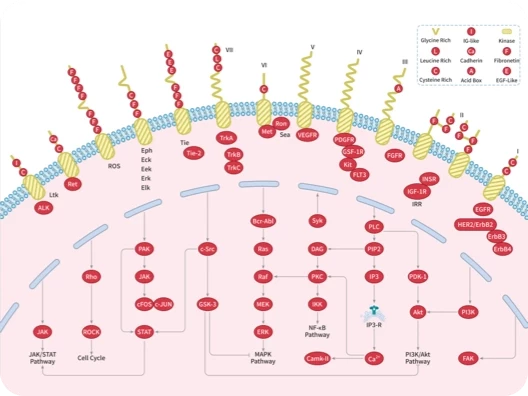
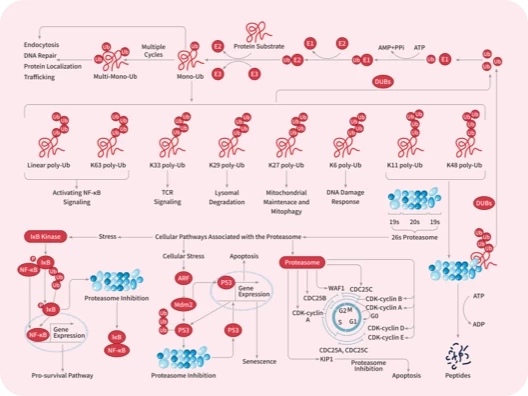
 您的购物车当前为空
您的购物车当前为空












 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询