购物车
 您的购物车当前为空
您的购物车当前为空
【TargetMol 明星分子】- Acetaminophen: 探索其在炎症调控与表观遗传中的多重作用机制
背景介绍:
Acetaminophen(T0065),CAS: 103-90-2,是一种广泛研究的化合物,主要通过抑制环氧合酶COX-1和COX-2发挥作用,其IC50分别为113.7 μM和25.8 μM。该抑制作用调节了COX信号通路,这一通路在前列腺素的生物合成中起关键作用,前列腺素作为脂质介质调控炎症、疼痛和发热等生理过程。
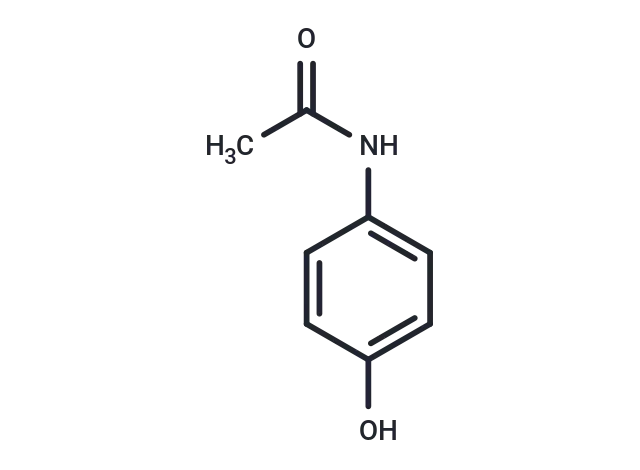
Acetaminophen 分子结构式
与传统非甾体抗炎药不同,Acetaminophen表现出显著的解热和镇痛活性,但抗炎作用较弱,这反映了其对COX同工酶及下游信号的复杂调控机制。除经典的COX抑制作用外,Acetaminophen还与组蛋白乙酰转移酶(HAT)相互作用,提示其可能通过调节组蛋白乙酰化进而影响基因表达,具有潜在的表观遗传调控功能。这种对酶活性的双重调节使Acetaminophen成为研究炎症信号与染色质重塑相互作用的重要工具。
在科研领域,Acetaminophen被用来探究COX酶在不同生理和病理状态下的功能动态,帮助阐明前列腺素介导的信号机制。其对COX-1和COX-2的差异性抑制为研究不同同工酶在细胞过程中的特异性作用提供了实验手段。此外,Acetaminophen对组蛋白乙酰转移酶活性的影响,为研究代谢信号与炎症信号如何在表观遗传层面交汇提供了独特视角,可能影响细胞应激反应相关基因的转录调控。Acetaminophen作为内源代谢物参与的代谢通路,进一步揭示了小分子如何调控酶活性及表观遗传状态的复杂网络。
综上所述,Acetaminophen作为一种多功能的生化探针,在实验研究中有助于解析COX依赖的前列腺素合成及组蛋白乙酰化的动态变化。其应用涵盖炎症、疼痛信号传导及表观遗传调控的分子机制研究,促进对细胞稳态及应答机制的深入理解。凭借其明确的抑制特性和通路调控作用,Acetaminophen成为研究COX酶及染色质调节因子复杂生物系统的重要试剂。[1,2]
文献分享:
2.1 Hepatoprotective Effect of Citral on Acetaminophen‐Induced Liver Toxicity in Mice

Acetaminophen (T0065) 在250 mg/kg剂量下使小鼠发生急性肝损伤,病理表现为血清中转氨酶和磷酸酶(AST、ALT、ALP和γ GT)活性明显升高,组织学检查显示肝脏出现严重的坏死区,伴有肝中央静脉充血、炎性细胞浸润和肝细胞空泡变性。Acetaminophen诱导的肝损伤与氧化应激相关,导致核DNA断裂,随后引发炎症反应,包括促炎细胞因子的释放和免疫细胞的活化。通过髓过氧化物酶(MPO)活性的增加,观察到中性粒细胞浸润增强,提示炎症细胞在肝损伤中的参与。此外,研究指出一氧化氮(NO)在Acetaminophen肝毒性过程中起重要介导作用。[3]
2.2 Lactobacillus fermentum Postbiotic-induced Autophagy as Potential Approach for Treatment of Acetaminophen Hepatotoxicity

Acetaminophen(T0065),也称APAP,可在HepG2细胞中诱导保护性自噬。当与后生元HV110共处理时,自噬诱导效应显著增强:LC3蛋白转化水平提升3倍,p62/SQSTM1蛋白降解增加1.5倍,且p62/SQSTM1与PINK1的mRNA水平均出现上调。p62/SQSTM1 mRNA表达升高提示NF-E2相关因子2(Nrf-2)被激活,而Nrf-2是调控抗氧化基因表达的关键转录因子。
氯喹处理可降低对Acetaminophen/HV110共暴露细胞的存活率,这一结果证明HV110诱导的自噬发挥细胞保护作用;但氯喹处理后,细胞存活率并未降至单用对Acetaminophen处理的水平,提示还有其他保护机制参与该过程。MTT实验与LDH实验结果共同表明,HV110能够缓解对Acetaminophen造成的细胞损伤。综上所述,Acetaminophen对Acetaminophen与HV110共联合作用时,可增强了HepG2细胞中自噬介导的保护反应,从进而减少了细胞的毒性损伤。[4]
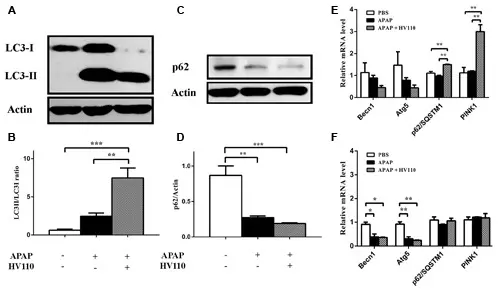
自噬与HV110保护效应之间的相关性
2.3 Acetaminophen Induces Human Neuroblastoma Cell Death through NFKB Activation

对Acetaminophen(T0065,本研究简称AAP)可诱导人神经母细胞瘤SH-SY5Y细胞发生死亡,其作用机制涉及激活内源性凋亡通路。当药物浓度为1~2 mM处理细胞72小时后,可观察到细胞活力显著下降。对Acetaminophen可促进促凋亡蛋白Bax向线粒体聚集,诱导线粒体释放细胞色素c,该过程在处理48小时时达到峰值;在药物处理18小时后胱天蛋白酶1被激活,随后在24~48小时内激活胱天蛋白酶3。处理48小时后可检测到典型的DNA片段化特征,证实细胞发生了凋亡。
此外,对Acetaminophen还可通过磷酸化激活NFkB p65亚基,促使其转位进入细胞核,上调IL-1β的表达上升,而IL-1β会进一步促进细胞死亡。使用肽类抑制剂SN50阻断NFkB的核转位,可有效抑制IL-1β生成及,减少细胞死亡。CYP2E1酶介导的Acetaminophen对Acetaminophen代谢在仅部分介导了该药物的细胞毒性中起部分作用,:抑制CYP2E1活性虽可显著减少但未完全阻止弱对Acetaminophen诱导的细胞死亡,表明但无法完全阻断该效应,提示未发生代谢的Acetaminophen原型对Acetaminophen也参与了细胞毒性作用。抗凋亡蛋白Bcl-xL过表达虽不会降低IL-1β产的生成,但能防止Acetaminophen可阻断对Acetaminophen以及IL-1β介导的细胞死亡。此另外,抗氧化剂SOD模拟物MnTBAP可完全阻断对Acetaminophen诱导的细胞死亡,说明活性氧在该过程中发挥了关键作用。
综上,对Acetaminophen通过Bax介导的线粒体途径、NFkB激活、IL-1β上调以及胱天蛋白酶激活,诱导神经母细胞瘤细胞发生凋亡。[5]
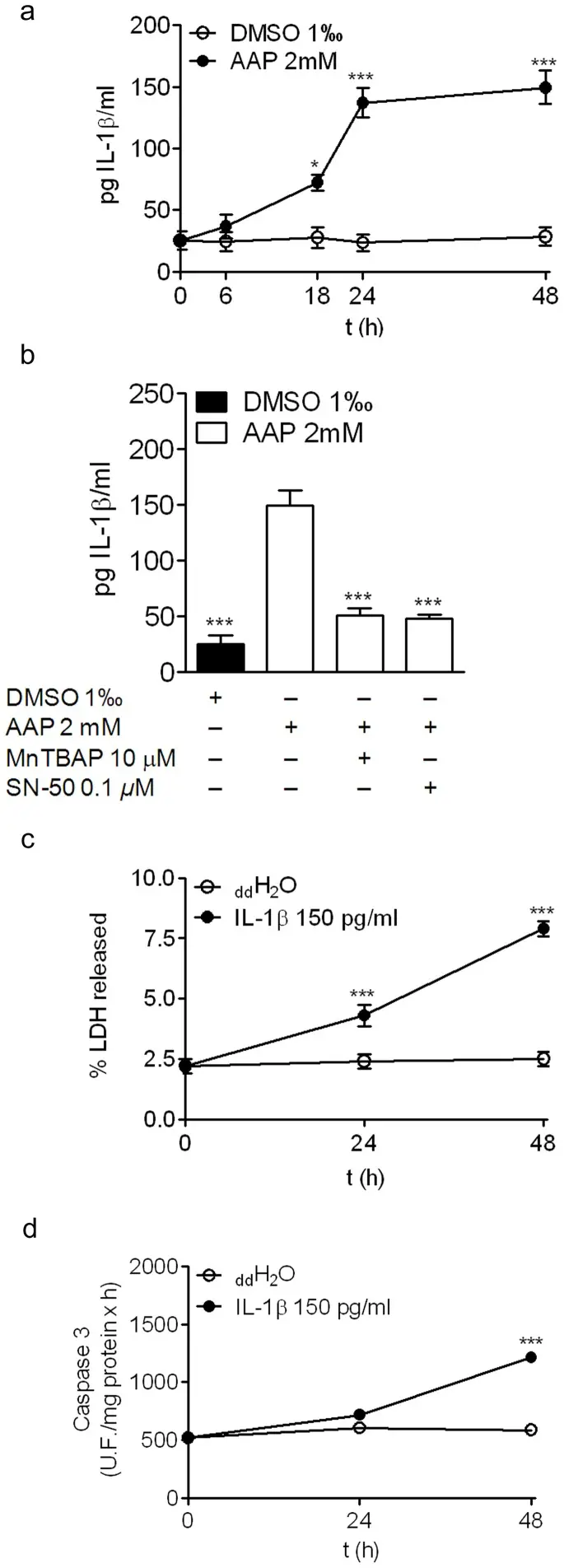
AAP诱导的IL-1β升高促进SH-SY5Y神经母细胞瘤细胞死亡
2.4 Glucocorticoids trigger muscle-liver crosstalk to attenuate acute liver injury and promote liver regeneration via the FGF6-FGFBP1 axis

Acetaminophen (T0065,本研究也称APAP) 诱导的急性肝损伤(ALI)引起皮质酮水平升高,激活骨骼肌中的糖皮质激素受体(GR),导致Fgf6基因转录受到抑制。该抑制作用激活了肌肉与肝脏之间的信号轴,从而保护肝脏免受损伤并促进肝脏再生。实验结果表明,特异性敲低肌肉中的GR会加重急性肝损伤并抑制肝脏再生,说明该信号通路的重要性。相反,Fgf6基因敲除的小鼠表现出肝损伤减轻和肝脏再生增强的改善效果。此外,肌内注射FGF6中和抗体能够逆转GR敲低所致的不良影响。这些结果共同表明,Acetaminophen诱导的ALI通过激活肌肉中的GR和抑制FGF6,调控了肌肉-肝脏间的通讯,从而促进肝脏的再生过程。本研究仅报道了Acetaminophen通过该途径间接促进肝再生的作用,未涉及该药物的其他效果。[6]
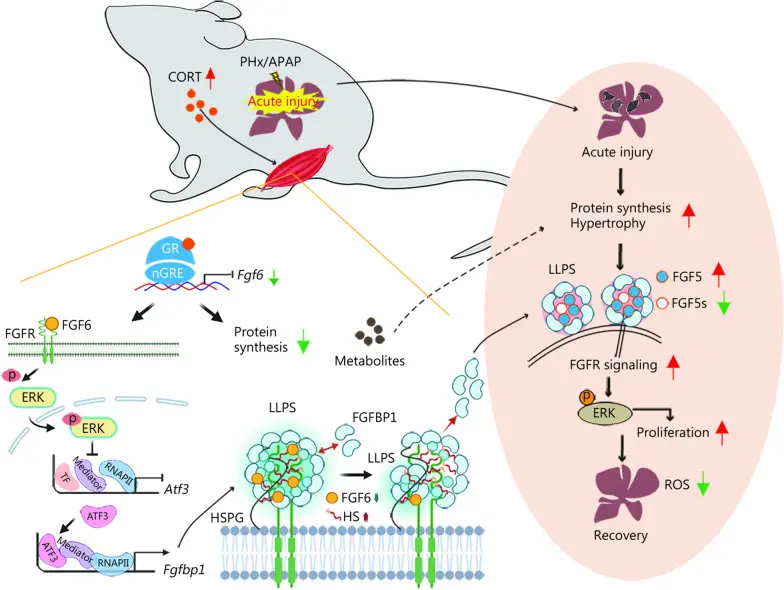
APAP诱导的ALI会提升CORT水平,激活肌肉GR以抑制Fgf6转录
2.5 Ferroptotic stress promotes macrophages against intracellular bacteria

本研究中发现Acetaminophen (T0065) 能够促进巨噬细胞内细菌的抑制作用。具体而言,作为铁死亡诱导剂,Acetaminophen加入巨噬细胞后,增强了巨噬细胞抑制细胞内细菌的能力。该效果与其他铁死亡诱导剂表现一致,表明Acetaminophen通过促进铁死亡应激,有助于巨噬细胞消除细胞内细菌。实验结果显示Acetaminophen支持巨噬细胞的防御机制,促进细菌细胞内死亡,从而减少被感染巨噬细胞中的活细菌数量。研究未报道或测试Acetaminophen的其他作用。[7]
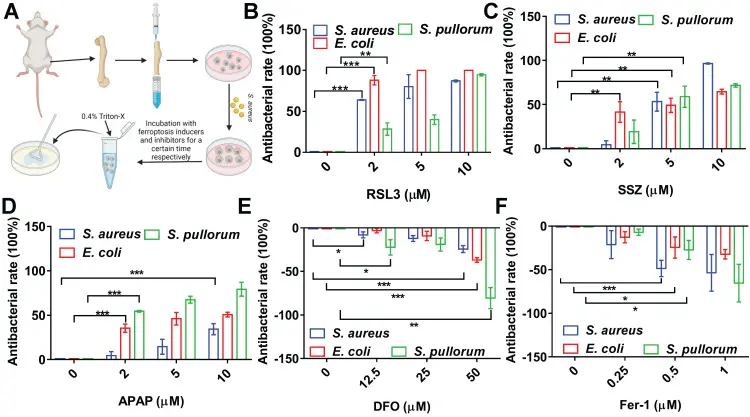
细胞内铁坏死样死亡有助于细菌在BMMs中的抑制
结语:
Acetaminophen主要通过选择性抑制COX-1和COX-2酶,调控参与炎症、疼痛和发热的前列腺素生物合成。此外,其与组蛋白乙酰转移酶的相互作用提示了通过调节组蛋白乙酰化实现表观遗传调控的潜力。文献综述显示,Acetaminophen诱导的肝毒性与氧化应激及炎症反应相关;与后生元共用时可增强肝细胞的保护性自噬;在神经母细胞瘤细胞中通过NFkB激活及线粒体途径诱导凋亡。同时,Acetaminophen调控肌肉-肝脏轴促进肝再生,并通过促进铁死亡应激增强巨噬细胞抗菌能力。未来研究可聚焦其酶活性与表观遗传双重调控功能,开发针对炎症疾病、肝损伤、神经退行性疾病及免疫增强的新型治疗策略。
Q&A
Q1: Acetaminophen主要通过什么机制发挥其药理作用?
A1: Acetaminophen主要通过抑制COX-1和COX-2酶,调节参与炎症、疼痛和发热的前列腺素合成的COX信号通路来发挥作用。
Q2: 文献中Acetaminophen对肝细胞有什么影响?
A2: Acetaminophen在小鼠中诱导急性肝损伤,伴随氧化应激、DNA断裂和炎症反应;在HepG2细胞中能诱导保护性自噬,且与后生元HV110共用时自噬增强,减轻细胞损伤。
Q3: Acetaminophen如何诱导神经母细胞瘤细胞死亡?
A3: Acetaminophen通过Bax介导的线粒体凋亡途径激活胱天蛋白酶,磷酸化激活NFkB p65并促进IL-1β表达,活性氧也参与其中,最终导致细胞凋亡死亡。
参考文献
[1] Hinz B, Brune K. Acetaminophen and cyclooxygenase inhibition: is there a cause for concern? Ann Rheum Dis. 2012;71(1):20-25.
[2] Chandrasekharan NV, Dai H, Roos KL, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99(21):13926-13931.
[3] Uchida N, Silva-Filho S, Cardia G, Cremer E, Silva-Comar F, Silva E, et al.. Hepatoprotective Effect of Citral on Acetaminophen‐Induced Liver Toxicity in Mice. Evidence-Based Complementary and Alternative Medicine. 2017;2017(1):.
[4] Dinić M, Lukić J, Djokić J, Milenković M, Strahinić I, Golić N, et al.. Lactobacillus fermentum Postbiotic-induced Autophagy as Potential Approach for Treatment of Acetaminophen Hepatotoxicity. Frontiers in Microbiology. 2017;8():.
[5] Posadas I, Santos P, Ceña V. Acetaminophen Induces Human Neuroblastoma Cell Death through NFKB Activation. PLoS ONE. 2012;7(11):e50160.
[6] Xu Y, Liu C, Chen Y, Li L, Xu B, You L, et al.. Glucocorticoids trigger muscle-liver crosstalk to attenuate acute liver injury and promote liver regeneration via the FGF6-FGFBP1 axis. Military Medical Research. 2025;12(1):.
[7] Ma R, Fang L, Chen L, Wang X, Jiang J, Gao L. Ferroptotic stress promotes macrophages against intracellular bacteria. Theranostics. 2022;12(5):2266-2289.
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询
版权所有©2015-2026 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4 | 沪公网安备 31010602006700号 | 沪(静)应急管危经许[2024]203441
| 沪(静)应急管危经许[2024]203441