购物车
 您的购物车当前为空
您的购物车当前为空
STTT:常驻热点!铜死亡&癌症&治疗应用!

铜死亡是今年来发现的一种独特的细胞死亡方式,对肿瘤抑制和治疗耐药性密切相关。先前T仔已经对铜死亡的特性表现做了详细解析“一文带你读懂热点 “ 铜死亡 ” ! ”,今天咱们再结合一篇有中南大学湘雅医院团队发表于 signal transduction and targeted therapy 期刊的综述 来看看铜死亡在癌症中的分子机制及治疗应用。

机体铜稳态调控
在生物体系中,铜通常以铜(II)和铜(I)两种氧化态存在。铜(I)主要存在于胞质内的还原环境中,而铜(II)则常见于胞外的氧化环境。由于铜具备较强的氧化还原能力,使其在生理过程中具有双重作用:
一方面,铜可作为多种酶的辅因子,参与电子转移;
另一方面,铜的过量积累可能干扰细胞代谢,进而导致细胞损伤甚至死亡。因此,必须通过严格调控的铜稳态系统,既保证酶类功能所需的铜供应,又防止铜的毒性积聚。
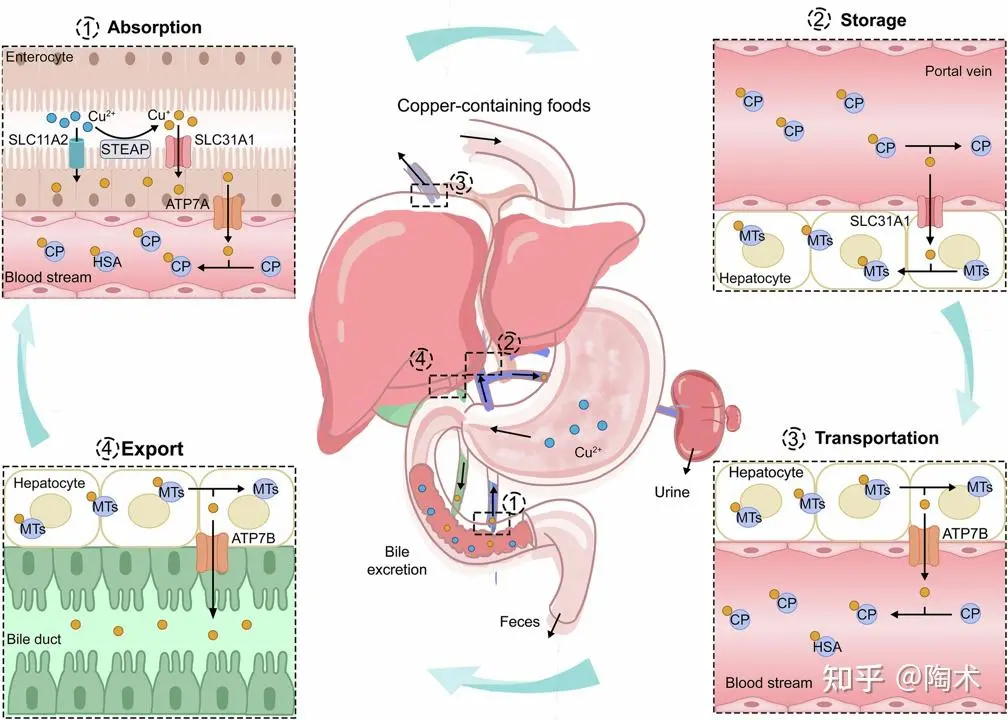
铜稳态的调控示意图
铜稳态包括铜的吸收、分布、利用和排泄等环节,这一平衡依赖于运输蛋白、铜伴侣蛋白和储存分子组成的调控网络,共同协调铜的摄入、细胞内运输、酶活性整合及最终排出。下面将分别从系统水平和细胞水平概述铜稳态的调控机制。
系统性铜稳态调控
系统性铜稳态包括肠道吸收、肝脏储存、全身运输和胆汁排泄等过程。膳食铜主要在小肠中被吸收,在此过程中,铜(II)通过STEAP家族成员被还原为铜(I)。铜(I)随后通过SLC31A1转运进入肠上皮细胞(肠细胞),少量也可通过SLC11A2进入。进入肠上皮细胞后,铜(I)与铜伴侣蛋白结合,并被运送至基底侧膜,通过ATP7A转运蛋白输出到血液中。
在血液中,铜(I)与可溶性的铜伴侣蛋白结合,主要是铜蓝蛋白(CP)。铜随后通过门静脉输送至肝脏,肝细胞通过SLC31A1从血液中摄取铜(I)。在肝细胞内,铜(I)可以被储存在金属硫蛋白 (MT)中,或通过ATP7B重新进入血液,以分配到其他组织。多余的铜在肝脏中被处理,并主要通过胆汁排出体外,这是铜排泄的主要途径。
细胞内铜稳态调控
在细胞内部,铜(I)可以被金属硫蛋白(MTs)和谷胱甘肽(GSH)螯合,形成一个动态的铜池,或与铜伴侣蛋白结合(包括CCS、COX17、CuL 和 ATOX1),并被运送至不同的细胞器中。
其中,CCS将铜(I)递送至超氧化物歧化酶 1(SOD1),帮助其将超氧自由基转化为氧,从而保护细胞免受氧化应激损伤。COX17则将铜(I)转运至线粒体内的SCO1和COX11,这对细胞色素氧化酶(COX)的组装至关重要。CuL可与胞质中的铜结合,并通过SLC25A3介导将铜输送入线粒体。ATOX1则将铜(I)递送至位于反式高尔基网络(TGN)的ATP7A/B。
当细胞内铜含量过高时,ATP7A/B会转移至囊泡结构并与质膜融合,将多余的铜排出细胞。此外,CCS和ATOX1也参与将铜运送至细胞核,这对于激活多种转录因子具有重要作用。
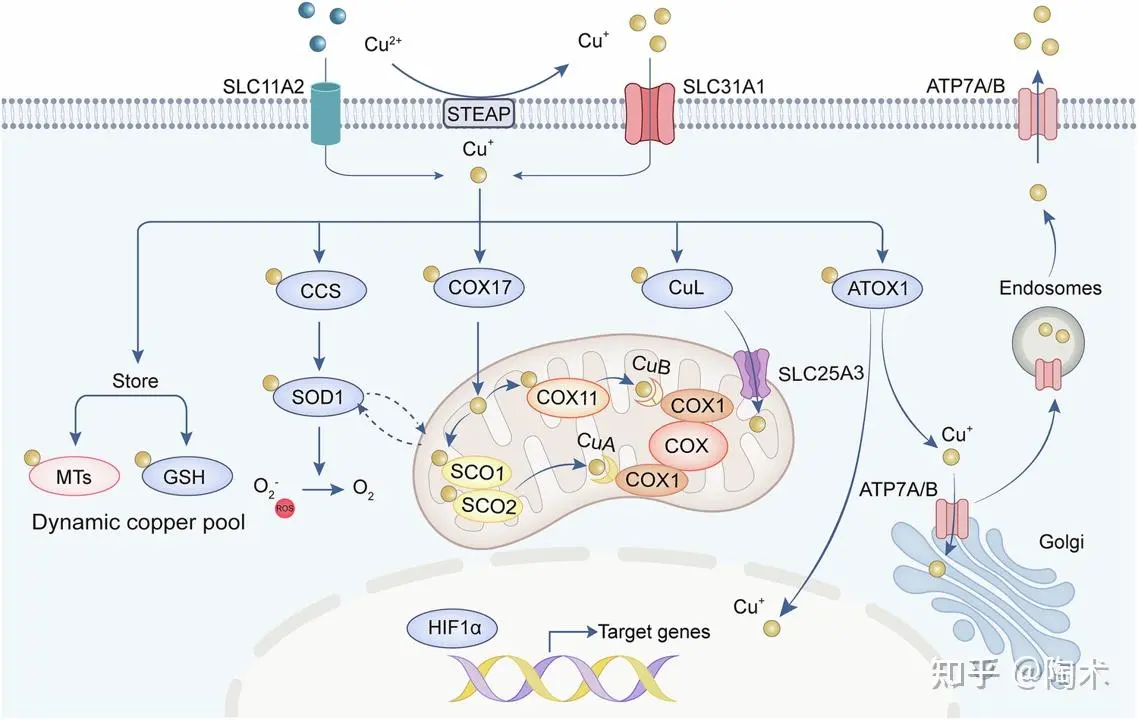
细胞内铜稳态调控示意图
铜在癌症中的作用
与正常组织相比,肿瘤组织对铜的需求会显著增加。其主要原因在于铜作为多种酶的辅因子,参与细胞能量代谢(如细胞色素c氧化酶,CCO)及抗氧化防御(如超氧化物歧化酶,SOD),从而满足快速增殖的肿瘤细胞对能量的高需求。
此外,由于铜具有较强的氧化还原活性,并可能与功能性大分子发生异常结合,因此在某些情况下也可能对肿瘤产生不利影响。因此,铜稳态的失衡可能在不同情境中对肿瘤起到促进或抑制的双重作用。
铜促进肿瘤发生的机制
铜能激活一系列与癌症相关的激酶信号通路,包括:
1. 致癌信号激活与自噬 调控:铜可与MEK1/2和PDK1结合,激活致癌信号通路;激活ULK1/2,或进入细胞核诱导铜结合蛋白CRIP2的降解,从而促进自噬;与p53蛋白结合并诱导其降解;通过抑制磷酸二酯酶PDE3B,促进脂解代谢,这些过程共同驱动“铜增生”(cuproplasia)的发生。
2. 血管生成:铜可直接结合或激活血管生成因子如血管生成素(ANG)和一氧化氮(NO),并与缺氧诱导因子HIF-1相互作用,增强NF-κB的活性,促进多种血管生成介质的表达。铜转运蛋白CTR1与VEGFR2之间形成的二硫键可激活VEGFR2信号通路,进一步促进肿瘤新生血管形成。
3. 促进肿瘤转移:铜可促进赖氨酰氧化酶(LOX/LOXL)和HIF-1α的表达,并通过正反馈机制协同增强肿瘤转移能力。铜还可与CD147结合,促进其自身聚集,进一步增强肿瘤细胞的转移性。
4. 免疫逃逸:铜可通过多条通路上调癌细胞中PD-L1的表达,从而抑制T淋巴细胞功能并诱导其衰竭,促进肿瘤的免疫逃逸。
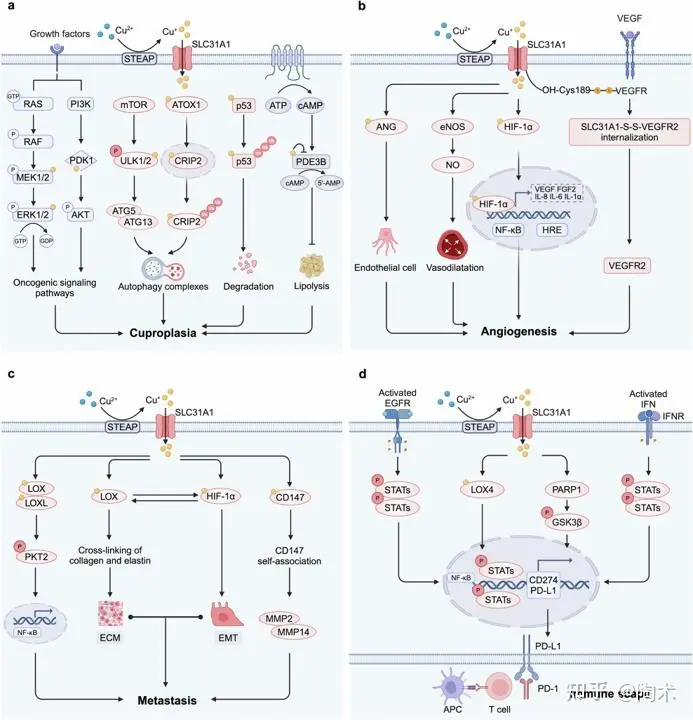
铜促进肿瘤发生的机制示意图
铜诱导肿瘤细胞死亡的机制
过量的铜可通过多种分子机制诱导细胞凋亡。包括:
1. 细胞凋亡(Apoptosis):铜主要通过诱导活性氧(ROS)生成、DNA损伤和蛋白酶体抑制来引发细胞凋亡。此外,铜可通过激活TP53靶基因的转录来诱导TP53依赖性凋亡,同时通过抑制核糖体合成和诱导核仁应激,触发TP53非依赖性凋亡。
2. 炎性细胞死亡(Pyroptosis):铜可通过诱导ROS产生和内质网(ER)应激,激活NLRP3炎症小体的形成,并通过GSDMD(Gasdermin D)激活在细胞膜上形成孔洞,从而诱导焦亡。
3. 程序性坏死(Necroptosis):铜毒性通过氧化应激激活TLR4/NF-κB信号通路,进而导致RIPK3和MLKL的磷酸化和寡聚化,诱导程序性坏死的发生。
4. 铁死亡 (Ferroptosis):铜可通过类Fenton反应和线粒体损伤生成ROS,引发脂质过氧化反应;同时铜可与谷胱甘肽过氧化物酶4(GPX4)结合并诱导其寡聚化,再通过TAXIBP1介导的自噬降解,从而促进铁死亡。
5. 自噬性细胞死亡(Autophagy-dependent cell death):铜可通过激活AMPK 、抑制mTOR ,或直接与ULK1/2激酶结合来启动自噬。铜还可上调自噬相关基因的表达,并激活转录因子TFEB ,促进自噬体和自噬溶酶体的形成,从而推动依赖自噬的细胞死亡过程。
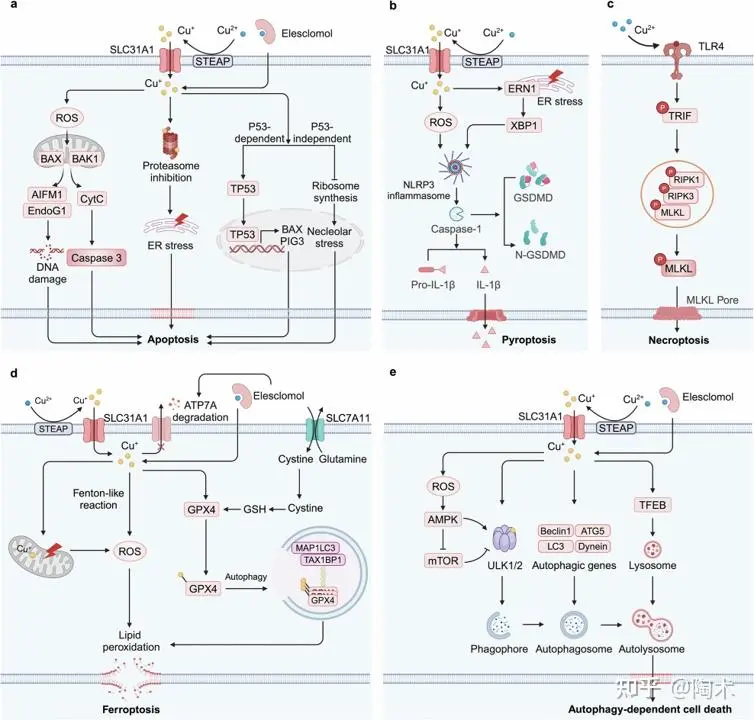
铜诱导细胞死亡的机制示意图
铜死亡的分子机制
铜死亡机制包括线粒体依赖性和线粒体非依赖性途径。
在线粒体依赖性路径中,过量的铜(II)进入线粒体后,被线粒体蛋白FDX1(铁氧还蛋白1)还原为更具毒性的铜(I)。FDX1 还可通过直接结合硫辛酸合酶(LIAS)并增强其与甘氨酸裂解系统蛋白H(GCSH)的相互作用,从而促进蛋白的硫辛酰化。铜(I)与这些硫辛酰化蛋白(如 DLAT)结合后诱导其聚集,并破坏铁-硫簇蛋白(Fe-S clusters)的稳定性,引发细胞应激反应,最终导致铜死亡。
在非线粒体依赖性路径中,二硫代氨基甲酸盐/铜(DSF/Cu)复合物可在细胞质中介导Npl4-p97复合物的聚集和构象锁定,从而抑制泛素-蛋白酶体降解通路,引起蛋白毒性应激(proteotoxic stress),也促成了铜死亡的发生
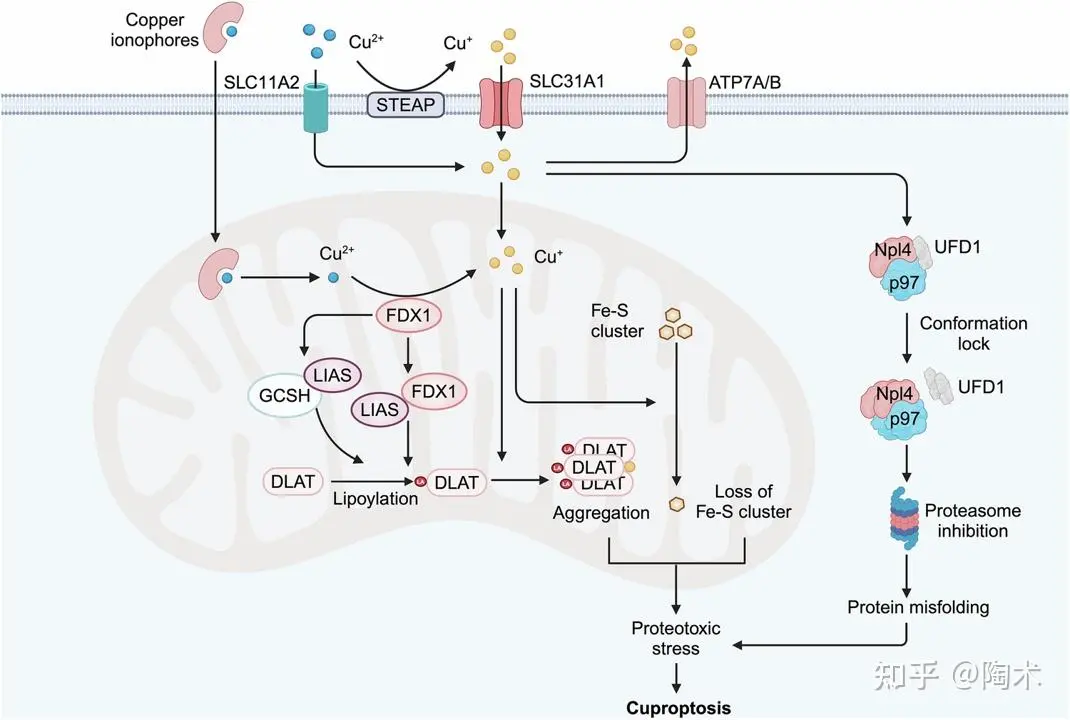
铜死亡机制示意图
铜与铜死亡的靶向治疗策略
铜与铜死亡机制在癌症中密切相关。细胞内铜水平受到严格调控,而铜稳态的失衡会显著抑制肿瘤进展。因此,通过消耗铜或引入铜以诱导铜死亡,为癌症治疗提供了新的策略。
当前,临床试验中用于癌症治疗的铜螯合剂/铜离子载体药物有:
铜螯合剂:
Tetrathiomolybdate (TTM) ,机制:抑制血管生成和转移
ATN-224 ,机制:抑制SOD1活性
D-penicillamine (D-pen or PCA) ,机制:抑制血管生成和转移
Triethylenetetramine (TETA/trientine) ,机制:抑制血管生成并促进细胞凋亡
Tetraethylenepentamine pentahydrochloride (TEPA),机制:抑制PD-L1表达
铜载体药物:
Elesclomol (ES) ,机制:诱导凋亡、铜死亡和铁死亡
Disulfiram (DSF) ,机制:诱导凋亡、铜死亡和铁死亡
Clioquinol (CQ) ,机制:诱导凋亡并抑制蛋白酶体功能
8-hydroxyquinoline (8-OHQ) ,诱导凋亡和空泡性细胞死亡(paraptosis),并抑制蛋白酶体功能
NSC319726 ,机制:诱导氧化应激和细胞周期阻滞
Pyrithione ,机制:诱导活性氧(ROS)产生和凋亡
欢迎有需要的老师私信了解详情噢~
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询
版权所有©2015-2026 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4 | 沪公网安备 31010602006700号 | 沪(静)应急管危经许[2024]203441
| 沪(静)应急管危经许[2024]203441