PD-217014 is a GABA analog. It inhibits (3)H-gabapentin binding to alpha(2)delta subunit of voltage-gated calcium channels in a concentration-dependent manner (K(i) = 18 nmol/l). PD-217014 possesses visceral analgesic activity.
Alaproclate is a selective serotonin reuptake inhibitor (SSRI).1,2 It inhibits depletion of serotonin (5-HT) induced by 4-methyl-α-ethyl-m-tyramine in rat cerebral cortex, hippocampus, hypothalamus, and striatum (EC50s = 18, 4, 8, and 12 mg kg, respectively).1 Alaproclate inhibits NMDA-evoked currents and depolarization-induced voltage-dependent potassium currents in rat hippocampal neurons (IC50s = 1.1 and 6.9 μM, respectively) and does not inhibit GABA-evoked currents when used at concentrations up to 100 μM.2 It increases sirtuin 1 (SIRT1) levels in N2a murine neuroblastoma cells expressing apolipoprotein E4 (ApoE4; IC50 = 2.3 μM) and in the hippocampus in the FXFAD-ApoE4 transgenic mouse model of Alzheimer's disease when administered at a dose of 20 mg kg twice daily.3 Alaproclate (40 mg kg) decreases immobility time in the forced swim test in rats, indicating antidepressant-like activity.4References1. Michael, G.B., Eidam, C., Kadlec, K., et al. Increased MICs of gamithromycin and tildipirosinin the presence of the genes erm(42) and msr(E)-mph(E) for bovine Pasteurella multocida and Mannheimia haemolytica. Journal of Antimicrobial Chemotherapy 67(6), 1555-1557 (2012).2. Svensson, B.E., Werkman, T.R., and Rogawski, M.A. Alaproclate effects on voltage-dependent K+ channels and NMDA receptors: Studies in cultured rat hippocampal neurons and fibroblast cells transformed with Kv1.2 K+ channel cDNA. Neuropharmacology 33(6), 795-804 (1994).3. Campagna, J., Soilman, P., Jagodzinska, B., et al. A small molecule ApoE4-targeted therapeutic candidate that normalizes sirtuin 1 levels and improves cognition in an Alzheimer's disease mouse model. Sci. Rep. 8(1), 17574 (2018).4. Danysz, W.P., A., Kostowski, W., Malatynska, E., et al. Comparison of desipramine, amitriptyline, zimeldine and alaproclate in six animal models used to investigate antidepressant drugs. Pharmacol. Toxicol. 62(1), 42-50 (1988). Alaproclate is a selective serotonin reuptake inhibitor (SSRI).1,2 It inhibits depletion of serotonin (5-HT) induced by 4-methyl-α-ethyl-m-tyramine in rat cerebral cortex, hippocampus, hypothalamus, and striatum (EC50s = 18, 4, 8, and 12 mg kg, respectively).1 Alaproclate inhibits NMDA-evoked currents and depolarization-induced voltage-dependent potassium currents in rat hippocampal neurons (IC50s = 1.1 and 6.9 μM, respectively) and does not inhibit GABA-evoked currents when used at concentrations up to 100 μM.2 It increases sirtuin 1 (SIRT1) levels in N2a murine neuroblastoma cells expressing apolipoprotein E4 (ApoE4; IC50 = 2.3 μM) and in the hippocampus in the FXFAD-ApoE4 transgenic mouse model of Alzheimer's disease when administered at a dose of 20 mg kg twice daily.3 Alaproclate (40 mg kg) decreases immobility time in the forced swim test in rats, indicating antidepressant-like activity.4 References1. Michael, G.B., Eidam, C., Kadlec, K., et al. Increased MICs of gamithromycin and tildipirosinin the presence of the genes erm(42) and msr(E)-mph(E) for bovine Pasteurella multocida and Mannheimia haemolytica. Journal of Antimicrobial Chemotherapy 67(6), 1555-1557 (2012).2. Svensson, B.E., Werkman, T.R., and Rogawski, M.A. Alaproclate effects on voltage-dependent K+ channels and NMDA receptors: Studies in cultured rat hippocampal neurons and fibroblast cells transformed with Kv1.2 K+ channel cDNA. Neuropharmacology 33(6), 795-804 (1994).3. Campagna, J., Soilman, P., Jagodzinska, B., et al. A small molecule ApoE4-targeted therapeutic candidate that normalizes sirtuin 1 levels and improves cognition in an Alzheimer's disease mouse model. Sci. Rep. 8(1), 17574 (2018).4. Danysz, W.P., A., Kostowski, W., Malatynska, E., et al. Comparison of desipramine, amitriptyline, zimeldine and alaproclate in six animal models used to investigate antidepressant drugs. Pharmacol. Toxicol. 62(1), 42-50 (1988).
5α-Androst-16-en-3α-ol is a steroid pheromone that has been found in boar testes and human male axillary sweat and has diverse biological activities.1,2It enhances GABA-activated currents in primary mouse cerebellar granule cells (EC50= 0.4 μM).25α-Androst-16-en-3α-ol (0.1-1 μM) increases the amplitude of GABA-activated currents in HEK293 cells expressing human α1β2γ2and α2β2γ2subunit-containing GABAAreceptors.In vivo, 5α-androst-16-en-3α-ol (5-10 mg kg) decreases immobility time in the forced swim test in mice. It increases time spent in the open arms of the elevated plus maze in mice, indicating anxiolytic-like activity, when administered at doses ranging from 30 to 50 mg kg. 5α-Androst-16-en-3α-ol protects against seizures induced by pentylenetetrazole or electroshock in mice (ED50s = 48.9 and 21.9 mg kg, respectively). 1.Brooksbank, B.W., Brown, R., and Gustafsson, J.A.The detection of 5α-androst-16-en-3α-ol in human male axillary sweatExperientia30(8)864-865(1974) 2.Kaminski, R.M., Marini, H., Ortinski, P.I., et al.The pheromone androstenol (5α-androst-16-en-3α-ol) is a neurosteroid positive modulator of GABAA receptorsJ. Pharmacol. Exp. Ther.317(2)694-703(2006)
CAY10787 is an oxysterol and a negative allosteric modulator of GABAAreceptors.1,2It reduces GABA-induced currents in HEK cells expressing α1β1γ2or α4β3γ2subunit-containing GABAAreceptors (IC50s = 1.5 and 1 μM, respectively).2CAY10787 (500 nM) reduces GABA-induced depolarization of peptidergic and non-peptidergic nociceptors, C-LTMRs, and cold thermosensors in isolated mouse dorsal root ganglion (DRG) neurons.In vivo, CAY10787 (2, 10, and 50 mg/kg) increases latency to nocifensive behaviors in the hot plate test in mice. 1.Hahn, M., Tang, M., and Subbiah, M.T.Cholest-3,5-dien-7-one formation in peroxidized human plasma as an indicator of lipoprotein cholesterol peroxidation potentialBiochim. Biophys. Acta1255(3)341-343(1995) 2.Niu, C., Leavitt, L.S., Lin, Z., et al.Neuroactive type-A γ-aminobutyric acid receptor allosteric modulator steroids from the hypobranchial gland of marine mollusk, Conus geographusJ. Med. Chem.64(10)7033-7043(2021)
Potent and selective inhibitor of the glial glycine transporter GlyT1b (IC50 = 6.9 nM). Displays negligible activity at GlyT-2, adrenoreceptors, dopamine, 5HT receptors and noradrenaline, dopamine, 5HT and GABA transporters (pIC50 in vivo. Chue et al (2013) Glycine reuptake inhibition as a new therapeutic approach in schizophrenia: focus on the glycine transporter 1 (GlyT1). Curr.Pharm.Des. 19 1311 PMID:23194655 |Brown et al (2001) Discovery and SAR of org 24598-a selective glycine uptake inhibitor. Bioorg.Med.Chem.Lett. 11 2007 PMID:11454468 |Williams et al (2003) Development of a scintillation proximity assay for analysis of Na+ Cl- -dependent neurotransmitter transporter activity. Anal. Biochem. 321 31 PMID:12963052
GABAA receptor agonist 2 (compound 4c) is a highly potent GABAA receptor agonist which exhibits anti-depressive properties in classical mouse models of depression, such as the FST and TST. By binding to the GABA binding site on the GABAA receptor, GABAA receptor agonist 2 produces GABAergic effects. Notably, Compound 4c holds promise for depression research [1].
Piperazine (2HCl) is gamma-aminobutyric acid (GABA) agonists and its major effects appear to be on the central nervous system. Piperazine was the anthelmintic with the greatest number of reports of toxicoses and suspected toxicoses in cats. Piperazine neurotoxicity in cats and dogs usually was manifested by muscle tremors, ataxia, and or behavioral disturbances within 24 hours after estimated daily dose(s) between 20 and 110 mg kg[1]. For di-substituted derivatives, ciprofloxacin was selected and hybrids were synthesized via substitution at piperazinyl-N4. The reaction of piperazinyl-NH of ciprofloxacin with selected drugs resulted in pronounced growth inhibition of standard as well as resistant bacterial strains[2]. The parent piperazine 6 was found to exhibit a reasonable activity toward the HeLa and MDA MB 231 tumor cell lines (IC50= 9.2 and 8.4 μΜ, respectively)[3]. Piperazine adipate (10 mM) causes mortality of A. galli and H. gallinae after a maximum of 30 min exposure, inhibits malate oxidation by 78%, and inhibits aldolase activity in both parasites. Piperazine adipate (10 mM) also inhibits cholinesterase activity by 96% in Ascaridia galli (A. galli) and 93% in Heterakis gallinae (H. gallinae). Piperazine adipate inhibits oxaloacetate reduction by 26% and 55% in A. galli and H. gallinae, resepctively[4].
SKF89976A is a potent GABA uptake inhibitor, which is selective for GAT-1 (IC50 values are 0.13, 550, 944 and 7210 μM for hGAT-1, rGAT-2, hGAT-3 and hBGT-1 respectively). SKF89976A inhibits transport current competitively (Ki = 7 μM) and transmitter-gated current non-competitively (Ki = 0.03 nM). It is able to pass the blood-brain barrier after systemic administration and is active in vivo.
GYKI 52466 is an allosteric AMPA receptor antagonist. It selectively inhibits AMPA-induced inward currents (IC50 = 7.5 µM) over NMDA- or GABA-induced inward currents in primary rat hippocampal neurons at 50 µM but also inhibits kainate-induced inward currents in the same cells (IC50 = 11 µM).2 GYKI 52466 (10 µM) reduces the amplitude of spontaneous excitatory postsynaptic currents (EPSCs) in the same cells. It increases the latency to seizure onset and reduces mortality in a rat model of generalized tonic-clonic seizures induced by 4-aminopyridine (4-AP) when administered at doses of 25 and 50 mg kg. GYKI 52466 (30 mg kg) prevents neuronal damage in the CA1 region of the hippocampus in a rat model of global ischemia-reperfusion injury induced by four-vessel occlusion.


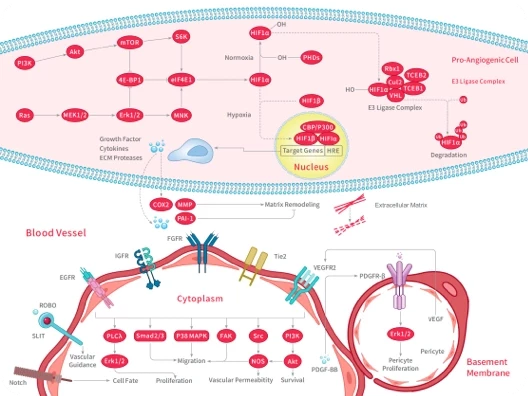
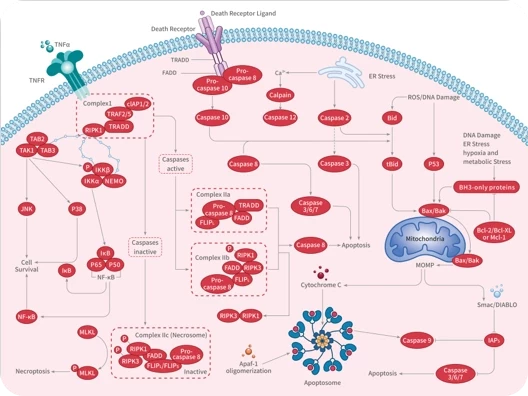
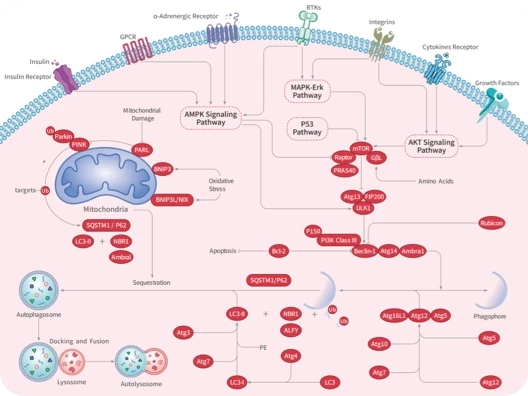

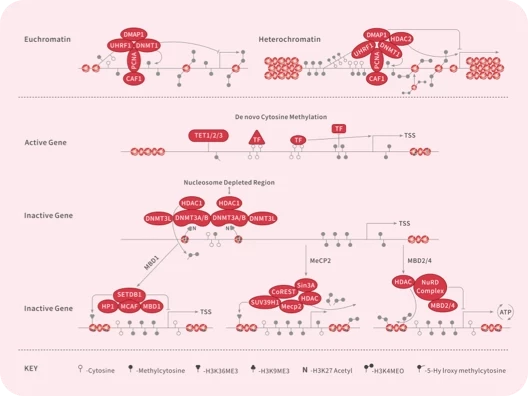
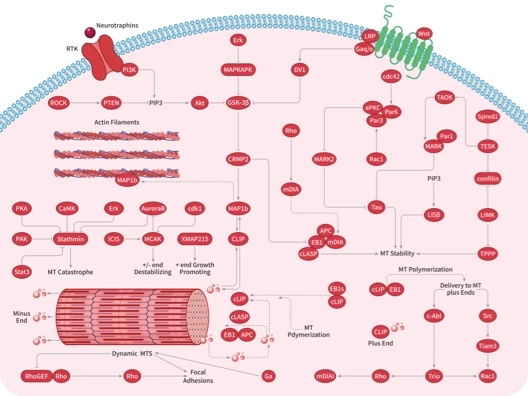
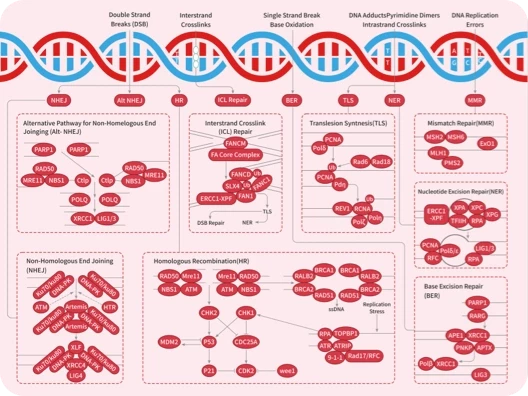
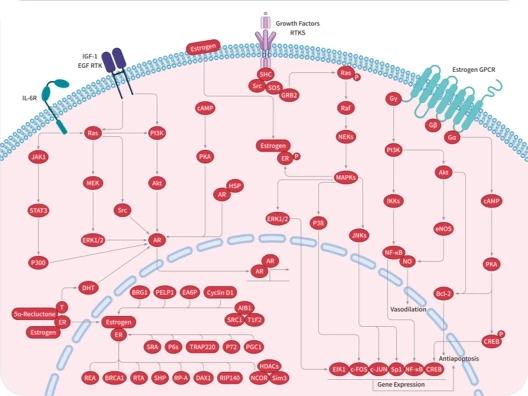
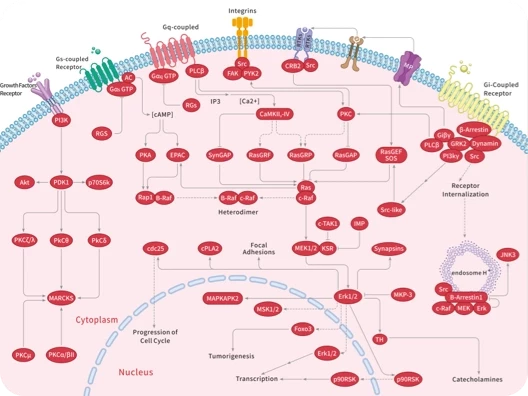
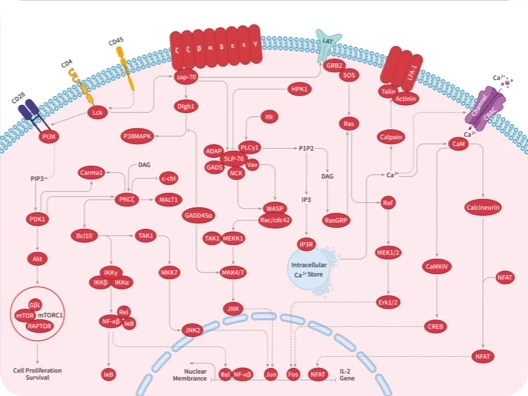
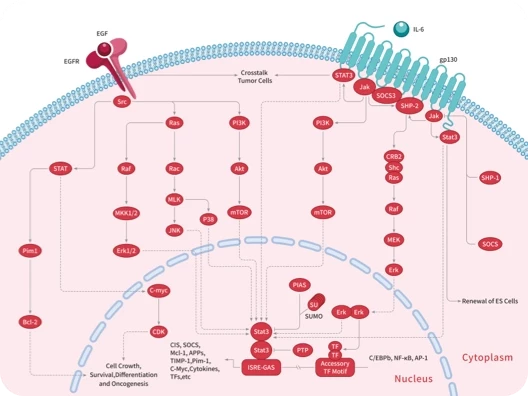
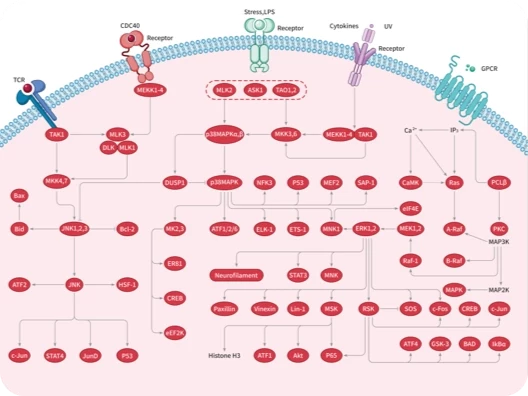
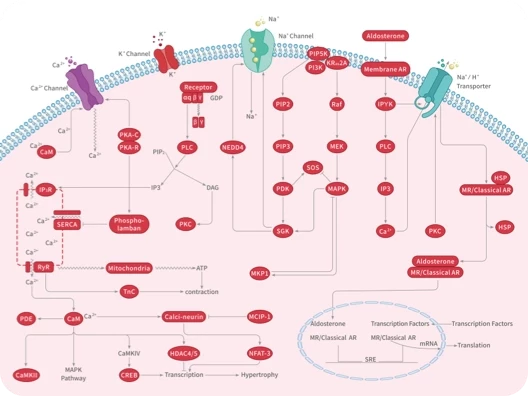

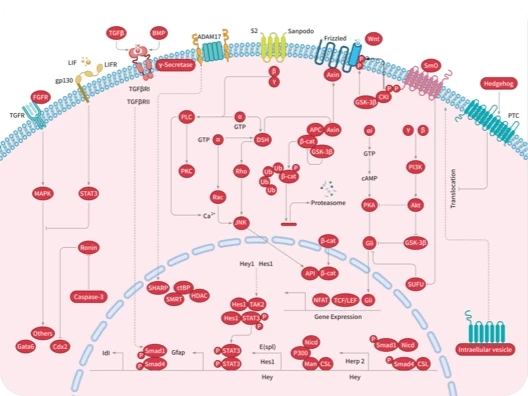
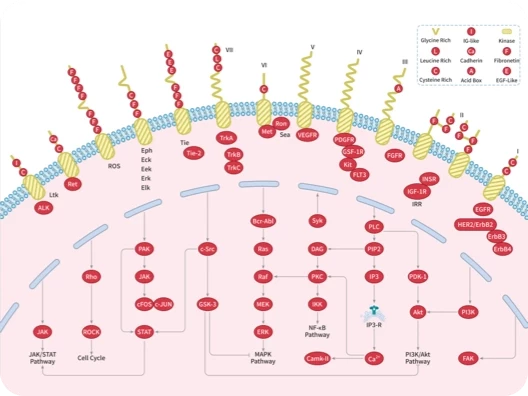
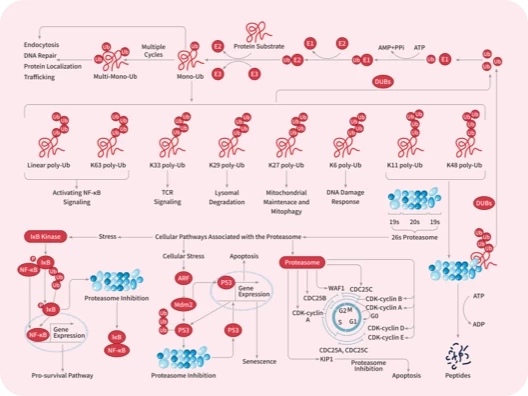
 您的购物车当前为空
您的购物车当前为空












 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询